Report
In 1992, the Life Extension Foundation introduced a melatonin supplement because of the broad-spectrum protective effects that this hormone had shown against age-related disease.1 Some of this research even suggested that melatonin supplementation may extend the human life span.2 Indeed, melatonin is so intricately involved in cell regulatory processes that scientists are now studying it as an adjunctive cancer treatment.3,4
These days, most people are likely to associate melatonin with a hormone that helps people sleep better or prevents jet lag.5,6 Few people realize that melatonin is a cancer-killing hormone7,8 that can enhance the human immune system,9,10 protect against the toxic side effects of chemotherapy4,11 and radiation therapy,12,13 and improve wound healing after cancer surgery.14,15 Even fewer are aware of ongoing clinical trials in which melatonin is being used to help cancer patients better manage their disease symptoms,16 improve their quality of life,17 and even increase their survival rates.4,11
Although the evidence demonstrating melatonin’s anti-cancer effects18 cannot be overstated, melatonin’s impact on cancer treatment remains largely unappreciated. This is likely because pharmaceutical companies have little to gain by advertising the anticancer efficacy of melatonin. In Europe, where melatonin is not even readily available, many clinical trials of melatonin have been conducted.19,20 US pharmaceutical companies, however, have shown little interest in even hosting, let alone funding, such critically important and potentially lifesaving clinical trials.
Life Extension Supports Clinical Trial
The Life Extension Foundation is collaborating with Cancer Treatment Centers of America on the first prospective, randomized clinical trial utilizing melatonin in patients with advanced lung cancer. Life Extension is providing, at no charge, high-dose melatonin and placebo supplements for this ongoing clinical trial, which will be the first in the US to examine the effect of melatonin supplementation therapy on quality of life and overall survival rates for patients with metastatic non-small-cell lung cancer.
Life Extension and the Cancer Treatment Centers of America hope to determine whether patients with advanced lung cancer suffer abnormal circadian rhythms and whether this affects their melatonin levels. The researchers hope that this trial will confirm the favorable clinical results documented by Lissoni and colleagues, whose recent European clinical studies indicate that in patients with metastatic non-small-cell lung cancer, five-year survival and overall tumor regression rates were higher in patients concomitantly treated with melatonin than in those treated with chemotherapy alone.4 While no patient treated with chemotherapy survived after two years, five-year survival was achieved in 3 of 49 patients treated with chemotherapy and melatonin. The researchers hope that similarly promising results could eventually convince mainstream medical practitioners to administer melatonin in combination with standard cancer treatment regimens to patients in earlier stages of cancer treatment.
Numerous, mostly European clinical studies already have examined melatonin’s therapeutic benefits to patients with different types of cancer who either did not respond to standard oncological therapies11,19 or were eligible only for supportive care (advanced cancer deemed untreatable by conventional standards).21,22 A literature search of the PubMed database found 806 publications on “melatonin and cancer.” Fifty-two articles were found concerning clinical studies utilizing melatonin in cancer patients. In this article, we will highlight and summarize some of the key studies concerning the use and mechanisms of melatonin as an adjuvant cancer therapy.
What Is Melatonin?
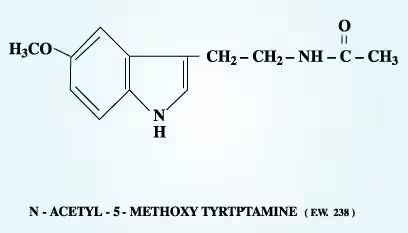
Melatonin (known scientifically as the indoleamine N-acetyl-5-methoxytryptamine) is a hormone with neurotransmitter modulatory activity.23 It is produced from the amino acid tryptophan in minute quantities by the pineal gland when the eyes detect no light (i.e., in darkness or blindness, or during sleep). Melatonin also is produced by the retina24 and, in vastly greater amounts, by the gastrointestinal system.25 In fact, 400 times more melatonin can be found in the gastrointestinal system than in the pineal gland or bloodstream,26 where levels typically range from 0.1 to 10 nmol/L. Melatonin receptors are present in central nervous tissues,27 peripheral tissues,28 and steroidogenic tissues,29 including myometrial tissues of both pregnant and non-pregnant women.SPAN class=wwwMagTextRefNumber>30 Maternal melatonin crosses the placenta.31
Melatonin levels peak during the night but also increase after eating,26 which partly explains why one may feel sleepy after a meal and why patients with advanced cancer who suffer diminished appetite or tissue wasting have been shown to have reduced levels of melatonin.32-34 Once produced, melatonin remains in the bloodstream only a short time, on average between 20 and 90 minutes.23,35 This is because melatonin is highly fat soluble (lipophilic) and somewhat water soluble (hydrophilic), enabling it to easily penetrate every cellular compartment (membrane, cytoplasm, and nucleus) and, as far as is known, every cell in the body.36 Melatonin’s amphiphilicity, or ability to both absorb and repel water—in conjunction with its ability to act as a weak preventive antioxidant,37 a weak metal ion chelator,38 and in certain circumstances, a direct free radical scavenger39—enables it to counteract oxidative stress within the chaotic tumor microenvironment.40
Melatonin’s Anti-Cancer Mechanisms
Melatonin can kill directly many different types of human tumor cells.3,41 It is a naturally produced cytotoxin, which can induce tumor cell death (apoptosis).7,42 In instances where the tumor has already established itself in the body, melatonin has been shown to inhibit the tumor’s growth rate.43,44 Melatonin exhibits natural oncostatic activity and inhibits cancer cell growth.45 In patients in whom cancer already has become a noticeable physical burden and produces overt symptoms, melatonin has been shown to alleviate numerous cancer symptoms46 and to inhibit development of new tumor blood vessels (tumor angiogenesis),47 which in turn inhibits the cancer from spreading further (metastasis).48 Melatonin can retard tumor metabolism and development by lowering the body temperature;35 it is a natural inducer of hypothermia. Furthermore, as an inducer of antioxidants49 and itself a weak preventive antioxidant,37 melatonin hinders tumor cells from participating in free radical damage to normal cells and consequently limits oxidative damage to DNA,40 lipids,50 amino acids, and proteins.40
In the unfortunate circumstance in which cancer has already overwhelmed the body’s innate cancer-fighting capabilities, including the anti-cancer activity of naturally produced melatonin (levels of which are reduced in most cancer patients), supplemental melatonin may be beneficial.17,43 Melatonin plays a critical role in the host defense system against cancer’s progression by activating the cytokine system,51 which exerts growth-inhibiting properties,10 and by stimulating the cytotoxic activity of macrophages and monocytes.52
Administration of supplemental melatonin has been shown to be beneficial even in the supportive care of advanced and end-stage cancer patients: it lessens tissue wasting and diminishes weight loss, fatigue, weakness, and depression;17,21,43,47,53 enhances immune function;10 improves wound healing;54 and improves quality of life and survival rates.4 Furthermore, melatonin improves common symptoms found in both patients with advanced cancer and those undergoing chemotherapy; it counteracts anemia55 and lymphocytopenia,14,21 stimulates platelet production,21 enhances appetite,16 and diminishes cancer pain56 (including bone pain) through its natural analgesic properties.57 These are substantial benefits considering that approximately half of all patients diagnosed with cancer die because of poor symptom management.58
Report
Melatonin and Cancer Surgery
In peri- and post-operative cancer surgery, melatonin may prove beneficial in wound healing through its natural anti-inflammatory properties.14,59 Melatonin reduces tissue destruction during inflammatory reactions60 by limiting hypoxia-reoxygenation-induced damage,61 scavenging free radicals, and reducing the upregulation of pro-inflammatory cytokines,60 such as the interleukins and tumor necrosis factor-alpha. Furthermore, surgery induces immunosuppression, which could adversely affect tumor-host interactions in cancer patients having their tumors surgically removed. As melatonin inhibits the activation of the acute inflammatory response, it may inhibit immunosuppression while contributing to an immune reaction against the tumor.14 Moreover, melatonin can reverse the perception of pain sensation (hyperalgesia) that is secondary to inflammation associated with wound healing.56
In cancer patients undergoing surgical removal of gastrointestinal tract tumors, preoperative neuroimmunotherapy with melatonin and interleukin-2 (IL-2) was capable of neutralizing the surgery-induced reduction in white blood cell counts (lymphocytopenia).14 Melatonin thus may prove to be beneficial to cancer patients who elect surgical removal of their tumors, by improving wound healing, inhibiting tissue damage, reducing pain sensation and weakness, counteracting reduced blood cell counts and anemia, and preventing immunosuppression.
Melatonin and Radiation Therapy
Radiation requires the presence of oxygen to generate free radicals to kill tumor cells. It is well established, however, that most human tumors are poorly oxygenated (hypoxic) because of blood perfusion and diffusion limitations,62 intermittent blood flow in the tumor microcirculation,63 and the occurrence of anemia in cancer patients (reduced hemoglobin indicates reduced oxygen levels).64,65 In fact, radiation therapy itself usually induces anemia, which is associated with a poor prognosis in cancer patients.66 Melatonin stimulates platelet production (thrombopoiesis)67 and has been shown to effectively treat cancer patients with low platelet counts and anemia.68
Moreover, melatonin has an anti-serotonergic effect, which means that it may block the inhibition of blood flow by serotonin.26 This consequently may increase blood flow and allow restoration of the microcirculation, which is compromised in the tumor microenvironment.69 Melatonin may improve the blood supply to the tumor, increasing tumor oxygen levels and thus increasing radiation-induced tumor cell death (by overcoming radio-resistance).70 In addition, melatonin is lipid soluble and can presumably cross the blood-tumor barrier as it does the blood-brain barrier.71 Melatonin may further increase the delivery of radiation (and chemotherapeutic drugs) to poorly oxygenated regions within the tumor microenvironment, consequently increasing the effectiveness of these anti-cancer modalities. Radiation, which frequently causes inflammation of the mucosa (mucositis), may substantially reduce melatonin levels in the body13 by damaging the mucosa of the gastrointestinal tract where melatonin is known to be localized.26
A radioneuroendocrine approach utilizing radiotherapy with melatonin supplementation in brain glioblastoma patients showed that the likelihood of survival at one year was significantly higher in those who received melatonin with radiotherapy versus radiotherapy alone.12 It recently has been suggested that melatonin may diminish the risk of hypoperfusion-induced cerebral ischemia.72 Therefore, melatonin supplementation may prolong the survival of patients undergoing radiotherapy.3 Melatonin also may provide relief from the inherent detrimental side effects of radiation treatment73 (including toxicity to the heart, kidneys, and nerves—cardiotoxicity, nephrotoxicity, and neurotoxicity, respectively), immune suppression, pain, anemia, fatigue, and sleep disturbances.12 Melatonin is a safe and effective facilitator of tissue repair processes, required for recovery from radiation-induced injury,74 and thus offers a promising co-treatment approach for patients undergoing radiation therapy for cancer.
Summary of Studies Using Melatonin
Lissoni’s Phase II Randomized Clinical Trial Results
One-Year Survival
| Tumor Type | Patient Number | Basic Therapy | Melatonin Dose | Melatonin | Placebo | Level Of Significance |
|---|---|---|---|---|---|---|
| Metastatic Non- Small-Cell Lung | 100 | Chemotherapy | 20 mg | 5-year survival 6% | 5-year survival 0% | N/A |
| Metastatic Non- Small-Cell Lung | 63 | Supportive Care Only | 10 mg | 5-year survival 6% | Under 1% | <0.05 |
| Glioblastoma | 30 | Conventional Radiotherapy | 10 mg | 43% | Under 1% | <0.05 |
| Metastatic Breast | 14 | Tamoxifen | 20 mg | 64% | 36% | <0.01 |
| Brain Metastases | 50 | Conventional Radiotherapy | 20 mg | 38% | 12% | <0.05 |
| Metastatic Colorecta | 50 | IL-2 | 40 mg | 36% | 12% | <0.05<0.05 |
| Metastatic Non- Small-Cell Lung | 60 | IL-2 | 40 mg | 24% | 19% | <0.05 |
Adapted from Life Extension (March 2002). Originally compiled by Cancer Treatment Centers of America.
Melatonin Dosage for Cancer Patients
While the optimal dose of melatonin for treating different types of cancer has not yet been established, the many clinical studies by Lissoni and colleagues have shown that doses of 10-50 mg of melatonin nightly are beneficial to cancer patients.
Those recently diagnosed with slow-growing or early-stage cancer may wish to consider supplementing with 3 to 6 mg melatonin nightly; the latter dose may be reserved for early-stage cancer patients who suffer from disturbed sleep patterns. Because most clinical studies have shown that patients with late-stage, advanced, or untreatable cancer, or those with cancer metastasis, benefit from supplementation with 20 mg of melatonin, such patients may wish to consider supplementing with between 6 and 50 mg of melatonin nightly, depending on plasma melatonin levels.
Physicians should be strongly encouraged to prescribe substitutional melatonin therapy to cancer patients with depressed melatonin levels.
Melatonin and Chemotherapy
Chemotherapy, through immunosuppressive and cytotoxic actions, exerts detrimental effects on patients’ physiological anti-cancer defense mechanisms. Melatonin, by improving immune status,52,75 has been shown to prolong survival and increase performance status in those undergoing chemotherapy. In conjunction with various chemotherapy regimens, melatonin has provided patients with a significant advantage over chemotherapy alone by increasing five-year survival rates, improving quality of life, and increasing the therapeutic effectiveness of many chemotherapeutic agents,76 while lessening or eliminating their negative and potentially detrimental side effects on normal healthy cells and tissues.4,77,78 Melatonin reduced chemotherapy-induced cardiotoxicity, neurotoxicity, nephrotoxicity, thrombocytopenia (reduced platelet counts), stomatitis (inflammation of mouth), and asthenia (weakness), and improved response in cancer patients.79,80
Melatonin should be seriously considered in combination with extremely toxic chemotherapy regimes—such as anthracyclines (adriamycin),81 cyclosporine, cytarabine,78 IL-2, cisplatin,55,79 5-fluorouracil,75,82 and methotrexate78,82—to reduce the incidence of their well-established side effects,80 which include but are not limited to mucositis and heart and liver toxicity.75 Melatonin recently has been shown to prevent methotrexate-induced liver and kidney toxicity in animals.83 It should be remembered that fasting reduces melatonin levels, typically within two days,84 suggesting that nausea, vomiting, and reduced appetite—side effects of chemotherapy—may reduce melatonin levels.
Melatonin and Chronotherapy
Because of the circadian rhythm dictated by the body’s melatonin levels, some types of chemotherapy work best if administered at an appropriate time of day, and are thus termed “chronotherapy.”3 The daily rhythm of melatonin exerts a “chronobiotic” effect and, as a circadian mediator, melatonin delivers the circadian signals to melatonin targets, including the internal body clock (in the suprachiasmatic nucleus).85 Chronotherapy is associated with maximum patient tolerability, tumor susceptibility, and attempts to improve the efficacy of treatment and the quality of patients’ lives. It takes advantage of asynchronies in growth rate between normal and tumor cells that are regulated by the circadian rhythm, thus minimizing damage to the patient and maximizing drug toxicity to tumor cells.
The growth of tumor cells may intrinsically follow a tumor-specific rhythm. It may be possible to modulate this rhythm by manipulating cancer patients’ melatonin levels.86 The local effect produced on the circadian clock could thus modulate the circadian rhythm.87 Slow-growing tumors could more likely be controlled by the patients’ circadian clock, whereas fast-growing or advanced-stage tumors may have altered circadian rhythms even though they are not temporally disorganized masses. High doses of melatonin are necessary to induce a phase-shifting effect on the circadian rhythm.88 Melatonin thus may have a unique ability to control the biological clock, consequently suppressing malignant growth and increasing the efficacy of cancer therapies. Chronotherapy has been shown to increase the survival time in children with acute lymphoblastic leukemia.89
Report
Melatonin and Hormonal Therapy
Melatonin levels in cancer patients have been correlated with tumor aggressiveness and progression.90,91 A high percentage of women with estrogen-receptor-positive breast cancer have low plasma melatonin levels.92 Conversely, melatonin inhibits human breast cancer cell growth45 and reduces tumor spread and invasiveness in vitro.48 Indeed, it has been suggested that melatonin acts as a naturally occurring anti-estrogen on tumor cells, as it down-regulates hormones responsible for the growth of hormone-dependent mammary tumors.93
Melatonin differs from the classic anti-estrogens such as tamoxifen in that it does not seem to bind to the estrogen receptor or interfere with the binding of estradiol to its receptor.94 Moreover, melatonin can increase the therapeutic efficacy of tamoxifen95 and biological therapies such as IL-2.96 How melatonin interferes with estrogen signaling is unknown, though recent studies suggest that it acts through a cyclic adenosine monophosphate (cAMP)-independent signaling pathway.93 It has been proposed that melatonin suppresses the epidermal growth factor receptor3 and exerts its anti-proliferative effects by inducing differentiation97 as proposed for melanoma cells.98
Regardless of the mechanism, in tumorigenesis studies melatonin reduced the incidence and growth rate of breast tumors and slowed breast cancer development.99 Furthermore, prolonged oral melatonin administration significantly reduced the development of existing mammary tumors in animals.100 In a metastatic hormone-refractory prostate cancer patient, oral melatonin (5 mg/day) induced disease stabilization for six weeks.44
Night Light, Melatonin, Meditation, and Cancer Incidence
Low levels of melatonin have been associated with breast cancer occurrence and development. Women who work predominantly at night and are exposed to light, which inhibits melatonin production and alters the circadian rhythm, have an increased risk of breast cancer development.101 In contrast, higher melatonin levels have been found in blind and visually impaired people, along with correspondingly lower incidences of cancer compared to those with normal vision, thus suggesting a role for melatonin in the reduction of cancer incidence.102
Light at night, regardless of duration or intensity, inhibits melatonin secretion and phase-shifts the circadian clock, possibly altering the cell growth rate that is regulated by the circadian rhythm.103 Disruption of circadian rhythm is commonly observed among cancer patients104,105 and contributes to cancer development and tumor progression.106 Cancer alters neuroendocrine system function in such a way that melatonin levels are lower in patients with non-small-cell lung cancer.107 Indeed, the circadian rhythm of melatonin is also altered in advanced gastrointestinal malignancies, such as colorectal, gastric, and pancreatic cancer, with respect to healthy humans.108
Deregulation of many circadian clock functions in the human body—including blood pressure, temperature, hormones, sleep-wake pattern, immune function, and digestive activity—has been used as an independent prognostic factor of survival time and tumor response for patients with certain metastatic cancers.109 The circadian rhythm alone is a statistically significant predictor of survival time for breast cancer patients.110
Several studies have shown that the circadian clock is involved in tumor suppression at the systemic, cellular, and molecular levels, and that cancer should no longer be treated as a local disorder. For instance, the circadian clock regulates the immune response. Disruption of circadian rhythms could therefore lead to immunosuppression, which could disrupt cancer cell immunosurveillance and promote tumor development; however, melatonin as a circadian mediator can target the endogenous clock86 and has been shown to inhibit immunosuppression.111
The phenomenon of light at night regulating melatonin levels may explain the spontaneous tumor regression reported to occur through meditation alone in cancer patients (when the eyes are closed and detect no light).112-114 The regular practice of meditation is associated with increased physiological levels of melatonin.115,116
Pharmacological doses of supplemental melatonin can resynchronize individuals shown to have disrupted circadian rhythms,36,117 such as night-shift workers.118 Thus, cancer patients with endogenously depressed melatonin levels may benefit from both meditation and substitutional melatonin therapy, to improve quality of life119 while potentially inhibiting tumor growth and spread.
Melatonin and Advanced Cancer
Numerous clinical studies by Lissoni and colleagues have shown that melatonin adjuvant therapy favorably influences the course of advanced cancer, leading to an improved quality of life and increased survival.17,21 In cancer patients with untreatable advanced solid tumors, melatonin significantly lowered the frequency of catabolic wasting (cachexia), weakness (asthenia), low platelet (thrombocytopenia), and white blood cell counts (lymphocytopenia) compared to patients who received supportive care only. Melatonin improved disease stabilization and increased survival percentages at one and five years.4,21
Melatonin deficiencies in advanced cancer patients may be due to altered circadian rhythm (disturbed sleep patterns), cancer-related anorexia-cachexia, and reduced food intake as melatonin is produced by the enterochromaffin cells in the gastrointestinal tract in response to feeding.25 Melatonin supplementation in turn increases appetite,26 diminishes tissue wasting,21,46 and restores sleep continuity in those with cancer.5,71,120 Administration of melatonin to patients with advanced cancer who have only short expected survival times results in some cases in disease stabilization and improvement of performance status.17,43,119
Melatonin Supplementation and Cancer
Extrapolating the reduced melatonin levels observed in aging humans121,122 to the cellular level, one might expect to find less melatonin at the cellular level in tumors32,107 compared to normal healthy cells if tumor cells “age” (because of their increased growth rate) more rapidly than normal healthy cells. The potentially lower melatonin levels in tumor cells could possibly be normalized by melatonin supplementation, which in turn would be expected to lead to a negative growth advantage in the tumor microenvironment and therefore inhibit tumor growth. Melatonin levels are depressed in individuals with cancers of different origins during the phase of primary tumor growth,110 whereas normal melatonin levels may be found when remission occurs.123
In summary, results of the numerous clinical studies in patients undergoing standard anticancer therapies—including chemotherapy, immuno-hormonal therapy, radiation therapy, and cancer surgery—suggest that individuals with cancer should consider melatonin supplementation under a physician’s supervision. While melatonin may be obtained through diet and enter the bloodstream, sources of natural melatonin production, such as food intake, gastrointestinal bacteria, and bile, may be reduced in cancer patients. Taken together, these factors, in conjunction with the short half-life of melatonin, provide a good basis for recommending melatonin supplementation as an adjuvant therapy for cancer.
With the current level of evidence on the multidisciplinary anticancer actions of melatonin, Life Extension believes that physicians should be strongly encouraged to prescribe melatonin to patients with certain tumor types on diagnosis or during early stages of tumor development. Continued research and clinical trials are imperative to further define melatonin’s role in the management of cancer’s physical and psychological symptoms and in the adjuvant treatment of cancer patients. Sadly, due to a lack of commercial opportunities, we are unlikely to see further clinical trials with melatonin in the US, other than those sponsored by foundations such as Life Extension.
Much remains to be learned about how practical therapeutics will be achieved with melatonin supplementation. Despite the many practical hurdles to the use of melatonin in the adjuvant treatment of cancer patients, particularly in the US, we remain hopeful that the overwhelming proof of melatonin’s efficacy will eventually drive its use in clinical applications.
Contraindications and Dosage
One study reported no contraindications to melatonin use.158 Because of unknown risk, pregnant and nursing women should take melatonin only under the close supervision of a physician or not at all.158 Some researchers have suggested that people with allergies, asthma, autoimmune diseases, and immune-system cancers, such as leukemia and lymphoma, should use melatonin with caution. Clinical studies have shown, however, that in leukemia and lymphoma patients, simultaneous administration of melatonin with IL-2 is beneficial in providing disease stabilization and in prolonging survival time.53
Who’s at Risk for Melatonin Deficiency?
- Apart from those confronted with cancer, melatonin-deficient individuals may include:
- the elderly, geriatrics, and those with age-related disease117,139,145
- shift workers, individuals exposed to light at night, and insomniacs39,146
- airline pilots, flight attendants, and frequent transcontinental flyers 6,147
- individuals with occupations involving high electromagnetic field exposure, including telephone or electric-line workers148
- those with pineal disease,149 pinealectomised individuals (those without a pineal gland),150 or those with suprachiasmatic nucleus involvement117
- quadriplegics151
- post-gastric26 or post-spinal-cord surgery patients151,152
- anorexics, bulimics, and those with poor appetite or subject to frequent vomiting136 or with irritable bowel syndrome, diarrhea, or ulcerative colitis25
- individuals undergoing total parenteral nutrition (intravenous nutrition),153 and those who fast chronically84
- those who suffer from delayed sleep phase syndrome, circadian rhythm variations, fibromyalgia, depression, or anxiety (treated by benzodiazepines)72,136, 154
- females who suffer cramping (uterine contractile disturbances) associated with menstruation,30 as melatonin has been shown to block prostaglandin production155 and depress spontaneous uterine contractility156
- individuals on blood pressure medication, such as beta-blockers, statins, or calcium channel blockers.157 Most medications prescribed to lower blood pressure also inadvertently reduce serum melatonin levels, including beta-blockers, calcium channel blockers, and calcium antagonists. An estimated 40% of individuals who take beta-blockers have sleep disorders that may be easily remedied by taking melatonin. It has been suggested that, in clinical trials, melatonin should be combined with statins to reduce the free-radical-mediated side effects of these cholesterol-lowering drugs.158
Studies in humans have shown melatonin toxicity to be remarkably low with no serious negative side effects even at high doses (3 to 6.6 g) administered over a period of 35 days.159,160 Nevertheless, minor reactions to melatonin supplementation such as sleepiness, vivid dreams, headache, abdominal pain, and nausea have been reported to occur occasionally in a small proportion of individuals.158 Excess melatonin production has rarely been seen except in polycystic ovary disease.161 More recently, an observational study found elevated serum melatonin levels in individuals with nocturnal asthma.162
Sources of Melatonin
Melatonin is present in all living organisms, including microalgae (green algae), bacteria, fungi, plants, small crustaceans (certain prawns and crayfish), fish, animals, and humans.163 Natural sources of melatonin, not standardized to provide a defined concentration, and with possible contaminants, also include medicinal plants such as feverfew (Tanacetum parthenium), St. John’s wort (Hypericum perforatum), and huang-qin (Scutellaria baicalensis),122,164 sometimes reaching levels of several nanograms per gram165 and possibly contributing to the therapeutic efficacy of the respective herbs.
High melatonin concentrations are found in seeds and some fruits such as tart cherries, bananas, and tomatoes.166,167 Melatonin also is found in food sources such as oats, rice bran, sweet corn, wheatgrass juice, and ginger. It has been shown that dietary melatonin (from plant sources) directly elevates the circulating level of melatonin in the body,168 as does smoking marijuana.169
The building blocks for natural melatonin production in the body include sufficient amounts of vitamin B6, vitamin B3 (niacinamide), and most important, the amino acid tryptophan, which is found in high quantities in foods such as nuts (soy, almonds, and peanuts,), seeds (pumpkin and watermelon), spirulina, beans, and tofu.
Who Should Supplement with Melatonin?
Melatonin is widely accepted for the treatment of sleep disorders and circadian rhythm disturbances,132,133 and is particularly effective for certain types of insomnia and sleep disorders in the elderly.134 Melatonin can facilitate the discontinuation of commonly prescribed sleeping medications, such as benzodiazepine therapy.135, 136 The “chronobiotic” effect of melatonin has been used to help re-synchronize individuals shown to have disrupted circadian rhythms (for example, blind people),88 in “delayed sleep phase” syndrome, night-shift work, and jet lag.118 In fact, the best clinical indication for melatonin is for alleviating jet-lag symptoms, particularly if taken at the bedtime of the arrival destination.118 In children, melatonin has been reported to be beneficial for treating colic, diarrhea, sepsis,50 and asphyxia.71,137
In advanced age, melatonin supplementation should be considered for the following reasons:- Melatonin production declines with age,121 and it has been shown that the aged have lower blood levels of melatonin. Elderly women have higher levels of melatonin compared to elderly men, which may be one reason why women live longer than men.
- Aged individuals with early neuropathological changes in the temporal cortex, where the Alzheimer’s disease process starts, have lower cerebrospinal fluid levels of melatonin.138
- The preventive antioxidant activity of melatonin may counteract free-radical-mediated degenerative diseases typical of the aged.139-141 Melatonin has been shown to be beneficial in the treatment of Alzheimer’s disease.142,143
- If aging is indeed a consequence of accumulated free radical damage, then the unique electro-reactive properties and intracellular distribution of melatonin should be advantageous in deferring the signs of aging.117
- Melatonin has beneficial effects on sleep disorders,144 which frequently afflict the aged.134
Melatonin Availability
Melatonin is available either as an over-the-counter drug or food supplement in the US, Argentina, Poland, and China. Although theLife Extension Foundation’s melatonin supplements are not registered as drugs, their purity has been certified and verified by an independent laboratory for the purposes of the ongoing lung cancer clinical trial. Unfortunately, this is not the case with many of the other readily available melatonin supplements, as certification is not mandated for food substances or additives.
For now, melatonin remains a relatively inexpensive nutritional supplement not yet controlled by the FDA or any other corporate or regulatory body. Interestingly, there has been mention of categorizing melatonin as a vitamin, which could be beneficial in compelling the medical establishment to finally recognize its importance. On the other hand, many pharmaceutical companies have started to patent therapeutic uses of melatonin: a Dutch company has patented a composition for intranasal melatonin administration, a French company has patented a melatonin agonist for the purpose of treating depression and sleep disorders, and an Israeli company has patented a method for treating or preventing symptoms of tardive dyskenisia by melatonin administration.
When to Take Melatonin
Melatonin should probably be taken 30 minutes to one hour before sleeping. Slow-release melatonin preparations may benefit those with various types of insomnia, as the oral bioavailability of melatonin is approximately 15%.170 Exposure to light at night, however, regardless of the duration or intensity of the light, can fully suppress or decrease melatonin levels.171
Reprint of a letter from Cancer Treatment Centers of America
November 6, 2003
Mr. William Faloon
Life Extension Foundation
3600 West Commercial Blvd.
Fort Lauderdale, FL 33309
Re: Status of Protocol CTCA 01-07: A Multi-Center Randomized, Double-Blind, Trial Evaluating the Chronotherapeutic Role of Melatonin in the Treatment of Stage IIIB and IV Non-Small Cell Lung Carcinoma
Dear Mr. Faloon:
On behalf of Cancer Treatment Centers of Americaâ, I’d like to take this opportunity to say “thank you” for your donation of the thousands of capsules of melatonin for our clinical trial investigating the effects of melatonin, given at the appropriate circadian phase, in patients with non-small cell lung cancer. Indeed, the support of the Life Extension Foundation has made this landmark study possible. I’d also like to take a moment to update you on the progress of our study.
Our participating centers have enrolled thirty-one (31) patients onto the trial since December 2002, and we are looking forward to a steady increase in the number of patients enrolled in the foreseeable future.
To implement this program, your donation of high quality 20 mg doses of melatonin saved us considerable expense and effort. As you know, both federal and local IRB regulations require that we obtain an independent assay of any chemical agent that will be used in a human clinical trial. To fulfill these requirements, randomly selected capsules of melatonin and placebos were sent for analysis (HPLC and gas chromatography) to the Roswell Park Cancer Institute (an National Cancer Institute-Designated Comprehensive Cancer Center). As expected, the Life Extension Foundation’s products achieved all specifications on purity and dose.
Your product donations are now helping us discover more about the role of melatonin in cancer treatment. Indeed, this will be the first prospective randomized cancer clinical trial in the United States to investigate: (1) whether lung cancer patients produce a nocturnal pulse of melatonin prior to therapy; (2) what fraction of patients with advanced lung cancer suffer abnormalities in their circadian activity/rest rhythm, and 3) whether or not melatonin therapy, delivered at the appropriate circadian phase, improves the quality of life and overall survival of NSC lung cancer patients.
Thank you once again for Life Extension Foundation’s continued and generous support. We look forward to keeping you updated on the progress of this landmark investigation.
Best Wishes,
Christopher G. Lis, MPH
Vice President
Research and Development
CANCER TREATMENT CENTERS OF AMERICA
Melatonin to fight Cancer and more…
by Scott Rollins | Jan 30, 2020 | Articles, Cancer, Conditions, Neurologic, Supplements
High dose melatonin has been shown to inhibit cancer through at least 8 different mechanisms. There are also numerous studies showing it helps prevent and treat Parkinsons and Alzheimers disease, heart disease and macular degeneration. It can protect against “chemo brain” and also from radiation induced damage.
Melatonin is incredibly safe at much higher doses than typically used to assist with sleep. It is not a soporific and most people can take it during the day without it causing sleepiness.
Melatonin dosing
- Prevention: Melatonin 180mg 30 minutes before bedtime
- Treatment: 60mg 1-2 capsule 3-6x/day
- 300mg 2 hours before PET/CT scan
- Only red light in bedroom at night to prevent sleep disruption
Melatonin Source
Not all melatonin is created equal. We recommend these sources.
Melatonin Max 60mg from Scientific Health Solutions is what we carry in office. For great savings with a little effort we recommend bulk powder from Pure Bulk where you may also purchase empty capsules and capsule maker to prepare your own melatonin capsules.
The pivotal role of melatonin in ameliorating chronic kidney disease by suppression of the renin–angiotensin system in the kidney
Abstract
Melatonin is a hormone produced by the pineal gland, predominantly at night, and plays a pivotal role in regulating the circadian rhythm as well as a variety of biological functions, including anti-inflammation, anti-oxidation, inhibition of sympathetic nerve activity, and preservation of endothelial cell function. The intrarenal renin–angiotensin system (RAS) is one of the most important contributors in the pathophysiology of chronic kidney disease (CKD) and hypertension, independent of the circulating RAS, due to sodium reabsorption and inflammation and fibrosis in the kidney. However, the relationship between melatonin secretion and intrarenal RAS activation has remained unknown. It has been recently shown that impaired nighttime melatonin secretion is associated with nighttime urinary angiotensinogen excretion, a surrogate marker of intrarenal RAS activation and renal damage in patients with CKD. Moreover, it has also been indicated that melatonin administered exogenously exercises antioxidant effects that ameliorate intrarenal RAS activation and renal injury in chronic progressive CKD animal models. As a result, the new roles of melatonin in suppressing RAS in the kidney via amelioration of reactive oxygen species have been clarified. Therefore, we review the relationship between melatonin and intrarenal RAS activation and indicate the possibility of a new strategy to suppress CKD, which is a risk factor for cardiovascular and end-stage renal diseases.
Relevant articles
Yenan Mo, Xina Jie … Xusheng Liu
BMC Complementary Medicine and Therapies | Open Access | 10 August 2021
Melatonin as a rational alternative in the conservative treatment of resistant hypertension
Fedor Simko, Russel J. Reiter & Ludovit Paulis
Hypertension Research | Open Access | 13 September 2019
Melatonin in Aging and Aging-Related Disorders
Abstract
Mammalian cells have multifaceted systems and structures to avoid accumulation of unfolded or misfolded proteins, the accumulation of which is a feature of aging and aging-related conditions. With a gradually increasing elderly population, aging-related pathologies such as neurodegenerative disorders, cancer, and cardiovascular diseases are becoming a growing economic, social, and personal problem. Few or no effective treatments are presented for aging-associated neurodegenerative conditions, which progress in an irreversible way.
Melatonin (MLT), a pineal hormone and an endogenous antioxidant, has numerous physiological functions in the brain, including regulating circadian rhythms, scavenging free radicals, preventing oxidation, and suppressing neuroinflammation. Clinical studies revealed that MLT levels are significantly reduced in patients with neurodegenerative diseases. Research proved that MLT prevents activity on neurodegeneration. As a chronobiotic, MLT can modify circadian rhythm. As a cytoprotective molecule, it prevents inflammatory injury in neurodegenerative diseases and aging.
This chapter discusses neurodegenerative diseases, which are the main risk elements of aging, antioxidant properties, and action of MLT, as well as MLT achievement in the pathogenesis of neurodegenerative diseases and its associations with the nine biological hallmarks of aging. The main physiological mechanisms of aging and their potential as targets of novel treatments for neurodegenerative diseases are also discussed.
Light, Water, and Melatonin: The Synergistic Regulation of Phase Separation in Dementia
Abstract
The swift rise in acceptance of molecular principles defining phase separation by a broad array of scientific disciplines is shadowed by increasing discoveries linking phase separation to pathological aggregations associated with numerous neurodegenerative disorders, including Alzheimer’s disease, that contribute to dementia. Phase separation is powered by multivalent macromolecular interactions. Importantly, the release of water molecules from protein hydration shells into bulk creates entropic gains that promote phase separation and the subsequent generation of insoluble cytotoxic aggregates that drive healthy brain cells into diseased states. Higher viscosity in interfacial waters and limited hydration in interiors of biomolecular condensates facilitate phase separation. Light, water, and melatonin constitute an ancient synergy that ensures adequate protein hydration to prevent aberrant phase separation. The 670 nm visible red wavelength found in sunlight and employed in photobiomodulation reduces interfacial and mitochondrial matrix viscosity to enhance ATP production via increasing ATP synthase motor efficiency. Melatonin is a potent antioxidant that lowers viscosity to increase ATP by scavenging excess reactive oxygen species and free radicals. Reduced viscosity by light and melatonin elevates the availability of free water molecules that allow melatonin to adopt favorable conformations that enhance intrinsic features, including binding interactions with adenosine that reinforces the adenosine moiety effect of ATP responsible for preventing water removal that causes hydrophobic collapse and aggregation in phase separation. Precise recalibration of interspecies melatonin dosages that account for differences in metabolic rates and bioavailability will ensure the efficacious reinstatement of the once-powerful ancient synergy between light, water, and melatonin in a modern world.
Keywords: melatonin; dementia; amyloid-β; ATP; adenosine; phase separation; infrared light; hydrogen bonds; viscosity; bioavailability
1. Introduction
Dementia is a neurodegenerative condition marked by varying levels of cognitive impairment [1], currently affecting approximately 46.8 million people around the world. It is estimated that 10 million people will develop dementia each year, and without approved pharmaceutical intervention to effectively target underlying causes [2,3,4], by the year 2050, healthcare spending attributable to dementia is projected to become a significant drain on resources representing 11–17% of total global healthcare spending [5]. Alzheimer’s disease (AD) is one of the most common causes of dementia [6,7] followed by vascular dementia (VaD) [8,9]. Together with Lewy body dementias [10] and frontotemporal dementia (FTD) [11], these major neurodegenerative disorders account for approximately 90% of all dementia cases [12]. Dysregulated aggregation of biomolecular condensates formed as a result of multivalent macromolecule interactions may underlie the common molecular mechanisms responsible for the development of all AD and non-AD dementia (nADD) [13,14,15].
An Alzheimer’s biomarker study performed within a defined population over a period of 15.7 years (maximum) found the absolute remaining lifetime risk for incident dementia to be significantly associated with elevated amyloid accumulation (hazard ratio 2.11; 95% CI 1.43–2.79). Even though 87% of the 4984 participants were diagnosed as cognitively unimpaired at enrollment, higher amyloid accumulation was a significant biomarker correlated with accelerated dementia progression [16]. Vascular risks are generally associated with the progression of VaD [8,9]. However, midlife hypertension and late-life amyloid-β (Aβ) deposition were found to be independently associated with increased dementia risk in 298 participants aged 45–64 in a study that spanned 30 years. The study was unable to identify evidence of synergy between vascular risk and Aβ deposition on a multiplicative scale in subjects with dementia, implying that unique molecular pathways may be involved in the development of dementia [17].
2. Aberrant Phase Separation Is the Fundamental Molecular Driver behind Dementia
In 2017, Banani et al. defined intracellular biomolecular condensates as cytosolic and nuclear micron-scale compartments not bound by membranes and formed via phase separation driven by multivalent macromolecular interactions [18]. These membraneless organelles (MLOs) are responsible for strategic cellular organization in response to changing environments including endogenous and exogenous stress [14,19,20,21]. MLOs are ubiquitously utilized not only by all eukaryotes, but also bacteria [22], and viruses which are now recognized as master architects of biomolecular condensates, using phase separation to form viral replication “factories” [23,24]. The disruption of phase separation in key cellular processes results in diseases including neurodegenerative disorders and cancer [25,26,27,28,29].
The forces that drive phase separation encompass simple density transitions in single-component fluid systems [30] to changes in macromolecule saturation levels in binary mixtures achieved via manipulating in vitro macromolecule expression levels, interaction energies, and inclusion of hydrotropes/surfactants [31,32,33,34]. Spontaneous or driven phase-separated biomolecular condensates are usually nonstoichiometric assemblies of multiple proteins and nucleic acids [35]. These multivalent macromolecules engage in site-specific interactions that conform to the “stickers-and-spacers” architecture [35,36,37], forming reversible crosslinks that may involve hydrogen bonds [38,39,40], ionic strength [41], cation-π, and π–π interactions [42] that fine-tune percolation thresholds that may further define phase separation processes [43,44,45,46].
The significant discovery by Kar et al. that fused in sarcoma (FUS) and other phase-separating RNA-binding proteins in the FET family, namely EWSR1 and TAF15, form reversible clusters of varying sizes in subsaturated solutions where phase separation was not observed [47], highlights the relevance of percolation without phase separation in phase transitions in vivo. The aggregation of FUS, EWSR1, and TAF15 are associated with neurological disorders and the three FET family RNA-binding proteins are widely expressed in most cell types [48,49]. Thus, the detection of FUS percolation clusters formed in subsaturated solutions and clusters that are coupled to phase separation in supersaturated solutions [47] offers additional insight in the aggregation of macromolecules in vivo where saturation concentration that can initiate phase separation has been questioned [50]. In this review, in order to accurately capture the concept that phase separation can be coupled to percolation as well as other phase transitions in vivo including the conversion to fibrillar solids [51,52], the term phase separation is employed in lieu of the more popular nomenclature of liquid–liquid phase separation (LLPS) which restrictively implies only viscous liquids are present in the coexisting phases [53].
2.1. Phase Separation of α-Synuclein into Amyloid Fibrils in Dementia
In 1992, Hardy and Higgins proposed that the deposition of amyloid fibrils in AD is the direct cause of cell loss, vascular damage, and dementia [54]. Continued research indicated that the AD disease process may be the result of the dyshomeostasis between the production and clearance of amyloid β-peptides (Aβ) [55,56]. The nomenclature committee of the International Society of Amyloidosis (ISA) defines in vivo amyloid fibrils as extracellular protein fibril deposits associated with 36 human amyloid proteins. Intracellular aggregates such as tau and α-synuclein (α-syn), which are present in all synucleinopathies and are the major component of Lewy bodies associated with Lewy body dementia and Parkinson’s disease (PD) [57,58], are excluded from this list [59,60]. However, the hallmark feature of amyloid fibrils is the self-association of soluble amyloid monomeric fibers into insoluble cross-β sheets [61,62,63], and both α-syn [64,65,66] and tau [67,68,69] have been reported to self-assemble into cross-β sheet structures.
Encoded by the SNCA gene on chromosome 4 [70], α-synuclein (αSyn) comprises 140 amino acids [71] with intrinsically disordered regions prone to fibrillization [72]. The aberrant self-assembly of physiological, soluble αSyn monomers into neurotoxic protein aggregates implicated in PD and other synucleinopathies [73,74] is now attributed to phase separation where macromolecular interactions trigger the irreversible liquid-to-solid transition into amyloid hydrogels containing oligomeric intermediates and cross-β-sheet fibrils [75,76,77,78,79]. The documentation of the conformational evolution of αSyn phase transitions has been successfully captured by solution and solid-state magic-angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopies [51], and the study and analysis of the material components as well as intermolecular interactions of protein molecules within αSyn condensates during phase separations were performed employing fluorescence recovery after photobleaching (FRAP) and Förster resonance energy transfer (FRET) techniques [80]. Since phase separation is an early event in αSyn aggregation, modulating phase separation and/or interfering with liquid-to-solid phase transitions during αSyn amyloid phase transitions become attractive molecular targets [81].
Phase transitions from soluble monomeric to insoluble β-sheet fibrils observed in medin employing C-direct detection NMR in combination with structural bioinformatics further supports the concept of phase separation as the common molecular pathway underlying not only AD, but also VaD [82]. Among the 36 amyloid proteins recognized by the ISA in 2018 [59], medin (AMed) is the most common amyloid found in the human body [83,84]—being an internal component of milk fat globule-EGF factor 8 (MFG-E8), also known as lactadherin—that is now associated with vascular Aβ in cerebral amyloid angiopathy (CAA) pathology [85].
Present in blood vessels of most adults over the age of 50, medin is cleaved from lactadherin to form insoluble amyloid aggregates [86,87] that co-localize with vascular Aβ deposits [88] to cause cerebrovascular dysfunction in aging mice and human subjects with VaD [89,90]. Cerebral arteriole medin is regarded as a novel biomarker for AD and VaD [91]. Even though the glycoprotein lactadherin has multiple, important physiological functions [92] including phagocytosis [93], angiogenesis [94], and mucosal repair [95], medin aggregates alter cellular homeostasis, causing microvascular endothelial dysfunction by inducing permeability via the formation of pores in lipid membranes that result in upregulated ionic current flow [96,97], a mechanism not dissimilar to how Aβ peptides form calcium ion channels in lipid bilayer membranes [98,99]. However, the conditions that trigger the cleavage of medin from lactadherin causing medin to self-assemble into pathogenic, insoluble fibrils remain unclear [82,83].
The self-recognizing aggregation of amyloid proteins is not limited to homotypic enrichment in one protein, but often also involves heterotypic interactions in condensates containing up to hundreds of proteins [100,101], and the outcome of amyloid aggregation is modified by these associated heterotypic interactions [102]. Aberrant phase separation resulting in delayed disassembly of stress granules (SGs) causes the formation of non-dynamic SGs that entrap and immobilize TAR DNA-binding protein 43 (TDP-43), rendering the protein insoluble in FTD pathogenesis [15]. Increasing understanding that phase-separating RNA-binding proteins such as FUS [47,103] and those that associate with tau [104] play important modulatory roles in the heterotypic interactions that can promote or suppress amyloid aggregation [102,105,106,107], warrants exploration of specific conditions that may trigger aberrant phase separation in RNA-binding proteins.
2.2. The Underappreciated Role of Hydrogen Bonds and Protein Hydration in Phase Separation in Dementia
Dementia-related neurodegenerative disorders are often associated with gene mutations that may cause the dysregulation of RNA-binding proteins responsible for the aggregation of pathological amyloid fibrils during phase separation [108]. The mechanisms reported mostly involve dysregulation in the low-complexity domains of proteins such as TDP-43 and FUS [103,105,109,110,111]. Low-complexity domains (LCDs) are generally regarded as universally disordered; however, LCDs can also adopt stable, structured conformations [112]. Therefore, aberrant phase separation observed in LCDs may involve other factors in addition to the dysregulation of intrinsically disordered regions that are essential in the promotion of phase separation. Intrinsically disordered proteins (IDPs) usually serve as necessary scaffolds that facilitate phase separation of biomolecular condensates [18,113,114,115] which can be tuned by controlling enthalpy, minimizing entropic costs in phase separation [116,117,118].
Intrinsically, phase separation is entropically unfavorable and driven predominantly by enthalpically favored protein interactions [119,120,121]. In addition to energetically favorable multivalent protein–protein interactions that offset entropic costs, variations in ions and salt concentration, pH, and temperature can result in thermodynamic changes in entropy–enthalpy compensation that regulate phase separation [122,123,124]. Phase separation in proteins such as Ddx4 [125] and hnRNPA1 [126] exhibiting upper critical solution temperature (UCST) cannot take place above a critical temperature at which the system remains homogeneous, whereas proteins exhibiting lower critical solution temperature (LCST) cannot phase separate below a critical temperature at which the system remains homogeneous [127]. Therefore, increasing temperatures can either stabilize or destabilize biomolecular condensates formed by phase separation [128], and variations in salt concentration and pH levels can further promote or disrupt phase separation [122].
Stress granules (SGs) are phase-separated membraneless organelles that are formed under endogenous and exogenous stress conditions; and persistent formation of stress granules may lead to fibrillization associated with neurodegenerative disorders [126,129]. Adjusting pH levels in solutions tunes both UCSTs and LCSTs that trigger phase separation [130,131]. Alterations in tightly controlled cytosolic pH not only affect the survival of yeast and other organisms, but also determine the material properties of phase-separated stress granule-like condensates that regulate stress responses [132,133]. A reduction in pH in yeast generates reversible condensates that dissolve upon restoration of neutral pH; whereas phase-separated condensates induced by heat in yeast can only be reversed with the help of chaperones [134]. Similarly, in lipid membranes, both pH and salt can increase or decrease critical temperatures that trigger phase separation [124].
In vitro elevation of salt concentrations produces either a dehydrating, salting-out (kosmotropic) effect that induces phase separation [135,136], or a hydrating, salting-in (chaotropic) effect that inhibits phase separation [137,138,139]. Classic interpretations of the Hofmeister effect where kosmotropic anions that remove water molecules from a protein’s hydration shell to reduce protein solubility, increasing potential for aggregation via electrostatic and hydrophobic interactions [140], and chaotropic anions that exhibit the opposite effect of increasing protein solubility, functioning as a hydrotrope preventing phase separation and aggregation [141,142] may not fully account for other relevant conditions including the reversal of the Hoffmeister effects in anions and cations [143], or the effect of pH on the aggregation of proteins relative to their isoelectric points (pI) [138].
At its pI of 4.7 pH, α-syn formed highly ordered, fibrillar structures even at low salt concentrations compared to other conditions due to favorable intermolecular energy interactions that compensated for the lack of salting-out effects in a low-salt environment [144]. The hydrophobic, hydrogen-bonded, Β-rich amyloid cores in α-syn are intrinsically disordered and participate in dynamic intermolecular energy interactions during fibril assembly and maturation [145,146,147]. As such, protein hydration exerts a distinct effect on the pathological aggregation of amyloid fibrils in dementia, as the hottest mutational spots are often located in residues that form protective hydrogen bonds but have lost their native protecting functions resulting in protein misfolding [148].
2.3. Hydration Water Activates Amyloid Aggregation and Regulates Oligomer Toxicity
The role of water hydrogen bond networks that hydrate protein surfaces in biomolecular systems is known to be active and dynamic [149,150,151,152], but its role in intracellular phase separation is often less understood. Hydrophilic residues are more hydrated than hydrophobic residues. Thus, entropy and enthalpy become the two fundamental thermodynamic driving forces in phase separation that provide the requisite energetically favorable decrease in free energy. Lum, Chandler, and Weeks postulated that the price for minimizing broken hydrogen bonds within interfacial hydration water compared to bulk is an increased enthalpic cost that scales with the surface area of the hydrophobic solute [153]. Therefore, the removal of hydration water into bulk (entropic) leads to increased protein concentration that facilitates enthalpically favored protein–protein interactions resulting in condensate formation [154,155].
In other words, desolvation or the release of water molecules from protein hydration shells into bulk water [156,157,158] create entropic gains that promote phase separation and fibril aggregation [136,159,160]. Tau proteins that phase separate from salting-out effects via increased salt concentration become dehydrated and mature into irreversible, canonical tau fibrils, whereas tau proteins in reversible condensates formed via electrostatically driven phase separation remain hydrated and do not mature into pathogenic fibrils with restricted water accessibility and increased micro-viscosity [135]. Mutational hotspots with structural defects that affect protein interactions in monomeric states can be regions with an immense propensity to aggregate if the exclusion or removal of water in those regions confer a high thermodynamic benefit [148].
In 1959, Walter Kauzmann proposed that hydrophobicity in protein hydration shells drives protein folding where protein hydration accumulates hydrophobic free energy and removing the water molecule from the hydration shell can supply the free energy required to drive protein folding [161]. This hypothesis remained largely controversial [162,163] until support from experimental evidence on protein hydration shells was published. When the original clathrate water hydration shell used by Kauzmann in 1959 was replaced by a dynamic one formed by van der Waals (vdW) attraction [164], it became clear that the structural differences between water molecules in hydration shells and bulk [165] contributed to changes in free energy produced in vdW attraction interactions that favored protein folding [166,167]. Furthermore, the fact that the addition of salt can tune the hydrophobic effect by breaking hydrogen bonds in hydration shells [168] and rearrange the hydrogen-bonding environment in interfacial waters [169], provides additional support for the role of dehydration in the formation of pathogenic amyloid fibrils.
Highly sensitive femtosecond time-resolved fluorescence spectroscopy revealed the presence of dynamically distinct, confined interfacial hydration water molecules with severely restrained mobility compared to bulk water [170]. The removal of these confined water molecules in the intrinsically disordered amyloidogenic NAC domain of a-syn changes the rate of intramolecular backbone reconfiguration to facilitate the formation of cytotoxic oligomers [171] via intermolecular associations involving chain desolvation, indicating the entropically favored removal of confined water molecules into bulk water [170]. Early studies found the aggregation of protofilaments from Aβ16-22 peptides was due to the hydrophobic collapse of protofilaments caused by water molecules being released [172,173]. Similarly, the aggregation propensity of Aβ1-40 was significantly elevated via escalating salt concentrations to enhance salting-out effects, with the implication of heightened protein–protein interaction energy and diminished hydrogen-bond strength [174,175]. Out of 3.45 hydrogen bonds formed by a water molecule, only 2.41 are considered “strong” hydrogen bonds. Per the hydrophobic effect, the ability to form hydrogen bonds directly affects the stability of protein where net stabilization at 1–2 kcal/mol can be provided by each intramolecular hydrogen bond [176].
Limited hydration in the interior of MLOs fosters a favorable environment for liquid-to-solid phase transitions observed in amyloidogenic aggregates that are often preceded by liquid-to-liquid phase separation [79,177]. During α-syn nucleation, limited hydration lowers the desolvation barrier and intermolecular hydrogen bond barrier. Thus, the simple removal of confined water molecules in the hydrophobic amyloid NAC domain in α-syn can easily breach high desolvation barriers that normally prevent aggregation of amyloid fibrils [178,179,180]. Furthermore, the level of protein hydration determines whether homogeneous or heterogeneous nucleation is selected as the primary aggregation mechanism, which further defines the type of amyloid polymorph generated as well as the cytotoxicity of the α-syn oligomers formed [178]. Unfortunately, reduced hydration may be an inevitable phenomenon associated with aging in the human brain.
During normal aging, even though total protein content in the normal aging brain can decline by 5–15% between the ages of 30 and 90 years, water-soluble protein content actually increases by 16–48%, providing a viable explanation for observations of significantly decreased water content in normal aging brain cells [181,182]. The fact that confined and “bridging” interfacial water molecules have limited mobility and exceptionally slow hydrogen-bond rearrangement compared to bulk water, respectively, [170,183] highlights the importance of the binding dynamics of interfacial hydration water around residues located in IDPs prone to phase separation under conditions of limited mobility and hydration [184,185]. Atomistic MD simulations revealed that during the growth of Aβ9-40 fibrils, the collective movement of confined interfacial water with reduced mobility provides the entropic energy for pathogenic fibril formation via the removal of 60–85 water molecules that concurrently supplies a dry binding interface between filament and monomer [186]. Consequently, the ability to manipulate the relative thermodynamics of hydrogen bonds [187] in interfacial water compared to bulk becomes an extremely attractive proposition in the regulation of protein aggregation in dementia.
2.4. The Synergistic Regulation of Hydrogen Bonds by Light, Water, and Melatonin
The International Union of Pure and Applied Chemistry (IUPAC) defined a hydrogen bond as “an attractive interaction between a hydrogen atom from a molecule or a molecular fragment X–H in which X is more electronegative than H, and an atom or a group of atoms in the same or a different molecule, in which there is evidence of bond formation” [188]. Although hydrogen bonding can affect important physicochemical properties including density, refractive index, and conductivity [189], due to a limitation of scope, this review is solely focused on the relevant associations between hydrogen bonds and viscosity [190,191] in the context of protein hydration in phase separation in dementia.
Interfacial water can exhibit viscosity 106 times higher than bulk water [192], and the breaking and forming of hydrogen bonds in water [193] can affect viscosity of interfacial water. Viscosity is measured in units of centipoises (cPs) [194], and viscosity can accurately indicate flow resistance in water and other solvents. Viscosity in interfacial water is increased by hydrophilicity and reduced by hydrophobicity, implying the strength of the hydrogen bond is critical to maintaining the integrity of the viscous phase in interfacial water [192]. Low-level microwaves, and other electric and electromagnetic fields (EMF) can restructure hydrogen bonding [195,196] where weakened or broken hydrogen bonds decrease viscosity [197,198] and the formation of stronger hydrogen bonds increases absolute viscosity [199]. Light is a form of electromagnetic radiation (EMR) [200], and plants are exposed to an entire spectrum of EMR from sunlight. However, plants only absorb visible light but reflect infrared light.
During canopy photosynthesis, visible light from the sun is absorbed and utilized while a directly proportional amount of infrared is reflected [201,202,203]. A tight, linear correlation exists between canopy photosynthesis and correspondingly reflected NIR in all types of plants examined, including well-watered crops, wetland vegetation, grasslands, and savannas, with different functions, structure, capacity, and even, soil conditions [204]. Surprisingly, or not, a higher level of greenness or presence of vegetation is associated with reduced risk for AD (20%, odds ratio 0.80; 95% CI, 0.75–0.85) and non-AD dementia (11%, odds ratio, 0.89; 95% CI, 0.82–0.96) [205]. However, subjects with dementia treated with UVB irradiation did not exhibit any of the greenness effect even though plants are exposed to both spectrums in sunlight [206].
Similarly, photobiomodulation employing visible red light (670 nm), non-visible near infrared (NIR, 800–1090 nm), and even far infrared (FIR, 3–25 µm) show encouraging results in the attenuation of symptoms associated with dementia including a reduction in Aβ deposition, size and number of plaque and fibril formation, clearance of misfolded proteins, increased ATP production and reduced ROS production, improved executive and cognitive functions, processing speed, memory performance, mood, energy, and sleep [207,208,209,210,211,212,213,214,215,216,217] (Table 1). The proposal that red and near-infrared wavelengths may promote melatonin synthesis in mitochondria via the pathway involving nitric oxide and enhanced activity of soluble adenylyl cyclase further bolsters the synergistic relationship between light and melatonin [218,219].
Table 1. A sample collection of popular wavelengths employed in photobiomodulation, starting from visible 670 nm to non-visible near- and far-infrared wavelengths, and their effects on various symptoms associated with dementia in animals and humans.
| Wavelength | Model/Cell Line/Device | Duration/Intensity | Results | Ref. |
|---|---|---|---|---|
| 670 nm | APP/PS1 AD transgenic mice/transcranial LED | 90 s (4 Joule/cm2)/day ? 20 over 4 wks | Attenuated cerebellar cortex A? deposition, fibril formation. | [207] |
| 670 nm | K3 tau, APP/PS1 AD transgenic mice/transcranial LED | 90 s (4 Joule/cm2)/day ? 20 over 4 wks | Neocortex and hippocampus of K3 and APP/PS1 mice showed reduction in tau/fibril formation and size/number of A?, respectively. | [208] |
| 670 nm | C57BL/6, transgenic 2576 mice/transcranial LED | 90 s (4 Joule/cm2)/day ? 20 over 4 wks | All mice showed reduced A? oligomer binding at CNS synapses. | [209] |
| 670 nm | h tau, 3xTgAD mice/transcranial LED | 90 s (4 Joule/cm2)/day ? 20 over 4 wks | Reduced toxic tau oligomers, improved memory deficits, upregulated clearance of misfolded proteins in both models | [210] |
| 808 nm | A?-treated microglia cells from health mice/diode laser | 5 min (9 Joule/cm2) | Exceeded control cell ATP production after 24 h by 155%, suppressed ROS production promoting neuronal survival. | [211] |
| 810 nm | 8 patients diagnosed with dementia/transcranial+transnasal LED | 20 min (pulsed at 40 Hz at 50% duty cycle), 3 times/wk for 12 consecutive wks | Significant score improvements in ADAS-cog (13.8%) NPI-FS (61.4%) compared to baseline�1. | [212] |
| 1060?1080 nm | 11 patients with dementia/transcranial LED helmet | 6 min (1100 LEDs pulsed at 10 Hz at 50% duty cycle)/day ? 28 consecutive days | Improved executive functioning in clock drawing, immediate recall, praxis memory, visual attention, and task switching. | [213] |
| 1060?1080 nm | 60 patients with mild to moderate dementia/transcranial LED helmet | 2 ? 6 min (23.1 mW/cm2)/day ? 8 consecutive weeks | Improved cognitive functions, auditory and verbal learning, processing speed, mood, energy, and sleep. | [214] |
| 1060 nm | 27 healthy participants aged 45+/transcranial LED helmet | 2 ? 6 min (12 mW/cm2)/day ? 28 minimum | Significant improvements in motor function, memory performance, and processing speed. | [215] |
| 1040?1090 nm | APP/PS1 AD double-transgenic mice/LED irradiation | 6 min (15 mW/cm2)/day ? 55 with a 28-day suspension after day 40 | Improvement in memory, spatial learning ability, and modest plaque reduction; suspension period indicated treatment effects were transient. | [216] |
| 500 nm/800 nm/3?25 �m | APP/PS1 AD double-transgenic mice/LED irradiation | 60 min (0.13 mW/cm2)/day ? 45 | FIR (3?25 �m) enhanced A? phagocytosis via increased ATP production and attenuated cognitive dysfunction compared to other wavelengths tested. | [217] |
The ability to increase adenosine triphosphate (ATP) production in mitochondria is one of the most widely accepted mechanisms behind the effectiveness of photobiomodulation in dementia and other health challenges [211,217,220,221,222]. The fact that both infrared light and melatonin increase ATP production, and the adenosine moiety of ATP which is structurally similar to melatonin is capable of solubilizing protein aggregation point to the existence of a most unexpected, dynamic relationship between NIR light and melatonin that is inextricably connected to the regulation of hydrogen bonds, viscosity, protein hydration, and protein aggregation (Figure 1). The following section will present what is currently known about molecular mechanisms that drive the synergistic relationships between light, water, and melatonin in the regulation of phase separation of pathological aggregates in dementia. In subsequent discussions, the term light refers to red and near-infrared wavelengths unless otherwise indicated.
3. Light, Water, and Melatonin: Ancient Synergies in a Modern World
The synergistic relationship between melatonin, water, and light may have originated billions of years ago when primitive unicellular organisms depended on this effective and precise synergy to modulate phase separation to control protein aggregation and associated biological effects. The efficacy of this synergy also provides a credible explanation for the immensely successful and rapid distribution of melatonin via horizontal gene transfer [223]. The discovery of the serotonin N-acetyltransferase (SNAT) gene responsible for the synthesis of essential melatonin substrate N-acetylserotonin (NAS) in archaea [224,225] firmly establishes the quintessential role played by melatonin in early primitive organisms that use phase separation as the fundamental driver for relevant biochemical and biophysical processes to support metabolism, replication, and survival [226,227,228,229,230,231,232].
Melatonin (N-acetyl-5-methoxytryptamine) was first isolated from bovine pineal gland in 1958 [233]. Since then, revelations from the study of melatonin led to a continuously expanding list of appellations that aim to describe its impressive yet often pleiotropic and contradictory behaviors. Melatonin is known as a hormone, an antioxidant, an anticancer agent, an antiviral, an autocoid, a chronobiotic, a hypnotic, an anxiolytic, a glycolytic, a sleep aid, a universal panacea, a biological modifier, and even a Higgs boson [234]. These nomenclatures are excellent illustrations of some of the broad-based metabolic effects achieved by melatonin as it regulates fundamental phase separation processes in living organisms. The role of melatonin in the regulation of phase separation in the context of neurodegenerative disorders, cancer multidrug resistance, and viral phase separation are clearly defined in several in-depth reviews [230,235,236]. Due to a limitation of scope, the reader may review these extensive discussions for a better understanding of molecular mechanisms employed by melatonin in the regulation of phase separation under different biological contexts. This review will focus on the presentation of known, relevant molecular mechanisms that facilitate and enhance the synergistic relationship between light, water, and melatonin in the regulation of phase separation in dementia.
3.1. Light, Water, and Melatonin: Viscous Relationships with Hydrogen Bonds
Water molecules confined in interfacial hydration water exhibit severely restrained mobility compared to bulk water [170]. The mobility of these water molecules is reduced by interfacial viscosities as high as 106 times that of bulk [192,237]. However, the viscosity of water constrained in extremely narrow spaces such as the interior of carbon nanotubes increases and decreases with increased and decreased diameters, respectively [238,239]. In carbon nanotubes with diameters below 20 Å, water stops behaving like bulk water with different boiling points, self-diffusion coefficient, and viscosity [238,240,241,242]. Even the mobility of water molecules in ultra-confined spaces is enhanced by reduced viscosity [239,243] which is facilitated by a reduction in hydrogen bonds.
In general, viscosity is increased by stronger intermolecular interactions that form more hydrogen bonds in water molecules [238]. During phase separation, the variation in internal micro-viscosity between tau droplets formed via homotypic and heterotypic associations can be as much as a 7-fold increase [244]. Systematic reductions in droplet micro-viscosity during biological aging may imply continuously evolving intermolecular interactions that shift droplet equilibrium, modifying aggregation potential that favor pathological outcomes [14,245,246]. Therefore, novel properties such as enhanced solubility, diffusion, and electron transfer in specially treated water molecules with lower viscosity and reduced/broken hydrogen bonds [247] may have distinctive effects on the modulation of aberrant protein aggregation in dementia.
Hydrogen bonds (HBs) can be reduced/broken by hot electron transfer when plain, deionized bulk water is allowed to flow through gold nanoparticles under resonant illumination. The water—known as plasmon-activated water (PAW)—created by this method exhibits features conspicuously different from bulk even at room temperature [247]. The reduced intermolecular hydrogen bonds in water molecules not only decrease viscosity, but also impart a higher degree of freedom in interaction that allows the formation of stronger intermolecular hydrogen bonding with hydrophilic solutes while enhancing the solubility of hydrophobic solutes [247,248]. Essentially, the elevated interactions with other molecules via increased free water molecules in PAW enhance the intrinsic activities of these molecules. Melatonin is known to dissolve poorly in water [249]; however, melatonin is able to form stronger hydrogen bonds in PAW resulting in enhancement of solubility between ~120% [248] to ~150% [250].
3.1.1. PAW Modulates Melatonin Hydrogen Bonding and Conformation
Melatonin has five distinct hydrogen bonding sites for water, forming up to five hydrogen bonds with water molecules simultaneously at varying strengths. Two of these hydrogen molecules from two water molecules can even reside indefinitely when they are coordinated with the O of the amide group due to the high degree of stability between the H-bond as indicated by Helmholtz free energy [251,252]. For melatonin, water can either be a H-bond donor or acceptor, depending on the site it is attached to. However, even one single water molecule attached to melatonin can change its conformational preference by modulating the relative energies of the conformations and the heights of the barriers that separate conformations, where strong H-bonds can produce substantial electronic frequency shifts. Furthermore, the relative abundance of the conformations can also be regulated by H-bonds, implying that preferential binding between specific sites and water molecules can produce conformational clusters with populations as high as 10 times over other species [252]. In bulk water, melatonin forms the strongest H-bond with its carbonyl O group, stabilizing its tendency to self-aggregate resulting in low solubility [253].
Melatonin prepared in PAW compared to bulk deionized water exhibited enhanced clearance of hydroxyl radical at 11.9% vs. 6.69%, respectively; its antiviral potency against dengue virus in infected human hepatocarcinoma cells is also enhanced, reducing infectivity by 14.7% vs. 20.6% in bulk [250]. Male Wistar rats subjected to chronic sleep deprivation (CSD) using the disc-on-water methodology [254] and treated with 10 mg/kg melatonin via intraperitoneal (IP) injection dissolved in PAW exhibited significantly better results in all parameters detected, including hepatic function and metabolic activity, than control (no treatment), CSD only, and CSD + melatonin dissolved in bulk deionized water groups [250]. It is plausible that when melatonin is dissolved in PAW, the intrinsic anti-inflammatory properties of PAW may also be responsible for molecular mechanisms that support/enhance melatonin’s antiviral and antioxidative features. Indeed, APP/PS1 transgenic AD mice treated with PAW showed improved memory function and reduced amyloid burden, potentially via anti-inflammatory and anti-oxidative effects, compared to age-matched wild-type controls [255,256]. There is no doubt that the anti-oxidative properties of PAW enhance melatonin’s intrinsic activities. However, the molecular mechanism involved is an unexpected, viscous one.
3.1.2. Reactive Oxygen Species Increase Viscosity
Hydrophilicity enhances viscosity in interfacial water at values up to ~106 times that of bulk due to an increase in ordering and hydrogen-bond dynamics [192]. The negative polarity of reactive oxygen species (ROS) is able to increase hydrophilicity and elevate viscosity. When the oxygen atom of one of the most reactive ROS hydroxyl radical (•OH) becomes highly negative and acts as a hydrogen bond acceptor, it can lower the reaction barrier stabilizing •OH bonding to water during the polar transition state. Thus, water and viscosity of water can modulate and stabilize the highly reactive •OH [257]. In bulk water, •OH forms three stable hydrogen bonds and a weaker hemibond with surrounding water molecules comprising its solvation shell [258].
In mitochondria, •OH is derived from superoxide radicals produced as a result of a one-electron reduction of oxygen (O2) from electron leakage during mitochondrial electron transport [259,260]. Simply stated, the presence of excess, unneutralized ROS can significantly elevate viscosity in these essential energy-producing organelles, negatively impacting mitochondrial functions and ATP production associated with pathological Aβ aggregation [261]. Hydrogen peroxide (H2O2)—a ubiquitous ROS with classical intracellular signaling functions at lower physiological levels [262]—is also produced in mitochondria from electrons leaked during mitochondrial electron transport activities [259]. Similar to •OH, H2O2 accumulation can increase matrix viscosity in mitochondria [263,264]. Furthermore, an NIR emissive fluorescent probe with a large Stokes shift detected significantly elevated viscosity and H2O2 levels in brain mitochondria of APP/PS1 transgenic AD mice compared to normal BALB/c mice [265].
3.1.3. Reduction in Viscosity and Hydrogen Bonds Enhance Melatonin ROS Scavenging
Melatonin is known for its ability to scavenge •OH and other free radicals [266,267,268,269,270] where one molecule of melatonin can scavenge two •OH radicals to produce the stable cyclic 3-hydroxymelatonin (3-OHM) metabolite [266]. However, the addition of only one water molecule that provides an H-bonding relay pathway significantly lowered the energy barrier in the tautomerization step to enhance the scavenging potential by melatonin [271]. The fact that melatonin prepared in PAW exhibit 78% increased effectiveness in •OH scavenging compared to bulk (11.9% vs. 6.69%) [250] implies that melatonin may adopt more favorable conformations that enhance its intrinsic activities as a result of stronger H-bonds formed in water with reduced viscosity and H-bonds compared to bulk.
In the context of aberrant protein aggregation in dementia, the signature reduction in viscosity and H-bonds in PAW inadvertently accentuates an unconventional but relevant perspective on the viscous relationships between light, melatonin, and ROS that surprisingly, or not, converge on the synthesis of ATP in mitochondria. In response to conditions that reduce ATP, budding yeast conserves energy by increasing cytosolic viscosity to slow cellular processes by reducing protein diffusion rates. Additionally, increased viscosity modulates phase separation, impeding the formation of stress granules and inducing aberrant phase separation to form aggregates that were not present in cells that could not elevate viscosity [272].
3.2. Light, Melatonin, and Viscosity in the Elevation of ATP Synthesis
Mitochondrial matrix exists mostly as interfacial water due to the density of proteins, and matrix water exhibits similar restrained mobility as interfacial water. Consequently, matrix water is significantly more viscous than cytoplasm [273]. The viscosity of the mitochondrial matrix is correlated with the respiratory state of the organelle that can affect not only signal transduction, but also how mitochondrial networks are organized. The abnormal elevation of viscosity in mitochondria results in dysregulation in metabolite diffusion that can cause aberrant phase separation resulting in malignancies associated with fatty liver, diabetes, atherosclerosis, accelerated aging, cancer, AD, and other neurodegenerative disorders. Therefore, the accurate detection and determination of mitochondrial viscosity can facilitate the understanding of molecular mechanisms behind various diseases associated with mitochondrial dysfunctions [265,274,275,276,277,278,279,280,281,282,283,284,285,286,287,288,289,290,291,292,293,294]. A fluorescent probe that can detect mitochondrial viscosity fluctuations was used for the first time in the successful, early diagnosis of liver and kidney injury in animal models [295], while other probes are employed to effectively distinguish normal cells from cancerous cells with distinct, elevated viscosity [296,297,298,299].
In HeLa cells, the average viscosity of mitochondria is determined to be ~62.8 cP [300], in stark contrast to the 2.04 +/− 0.49 cP obtained for HeLa nucleoplasm viscosity which is already higher than that in HeLa cytoplasm [301]. Furthermore, treatment with pharmaceuticals, such as monensin and nystatin, can further drive matrix viscosity up to 90.5 and 109 cP, respectively [300,302]. Mitochondria of HeLa cells under oxidative stress generate a tremendous amount of ROS [303]. Therefore, ROS such as •OH, which is naturally produced during mitochondrial respiration, and excess oxidative stress, can potentially increase matrix viscosity from its hydrogen-bonding interactions with water molecules. Dual-targeting fluorescent probes are developed to easily identify viscosity changes in mitochondria in the presence of specific free radical species [304]. Mechanistically, increased viscosity in the matrix can result in the lower production of ATP catalyzed by the ATP synthase.
3.2.1. Efficiency of ATP Synthase Is Modulated by Viscosity
The mitochondrial ATP synthase (F0F1) is a rotary motor enzyme with a proton-driven F0 motor that is embedded in the inner mitochondrial membrane and is connected to the ATP-driven F1 motor that protrudes into the mitochondrial matrix [305,306,307]. The higher viscosity of the medium can slow down the rotation of the F1 motor to reduce ATP synthesis not only in mitochondria [308] but also chloroplasts [309]. While ATPase turnover rates are more effective when detected by probes designed with lower viscous drag [310], viscous drag can dramatically slow the rate of rotation to 3% of the enzyme turnover rate in Escherichia coli [311].
Nonetheless, 100% efficiency of the F1 rotor can theoretically be achieved if the 120° power strokes rotate at a constant angular velocity [312]. However, power stroke and dwell duration are easily modified by viscosity. Viscous loads applied to the ATP F1 motor of E. coli can cause the increase in the duration of the 120° power stroke that is correlated to a 20-fold increase in the length of the dwell. Thus, the power stroke velocity is limited by the viscous load on the motor, and consequently, increases in transition time are the direct result of increases in viscosity and not from inhibition of the ATPase by other means [313]. A deeper analysis of viscosity sensitivity showed that viscous drag on rotations of the γ-subunit in the F1 motor [314] can cause variations of more than 5000-fold by using a variety of rotation probes [315].
3.2.2. The 670 nm Wavelength Elevates ATP Production in Mitochondria
The benefits of photobiomodulation employing the 670 nm wavelength for dementia and other neurodegenerative disorders are extensively documented (Table 1). Even though improved ATP production and reduced ROS production are associated with the use of 670 nm irradiation, the exact mechanism responsible for these effects remains controversial. Experimental works employing 670 nm report the reduction in inflammation via increased expression of cytochrome C oxidase (COX) in an age-related macular degeneration mouse model [316]; increased COX expression and ATPase activities in Wistar rats exposed to suppressive effects of fluorescent light [317]; significantly elevated ATP production in aging mouse retina via increased COX expression [318]; and the restoration of neuronal ATP and prevention of apoptosis induced by potassium cyanide—an irreversible inhibitor of COX [319]. Therefore, benefits from photobiomodulation, especially the enhancement of ATP production and mitochondrial function, are generally believed to be associated with the involvement of COX via increased COX expression and activities.
COX, or complex IV [320], is the fourth enzyme that catalyzes the transfer of electrons from ferricytochrome C to oxygen in the mitochondrial electron-transport complexes, and COX is highly susceptible to inactivation by oxidative damage induced by ROS including •OH and 4-hydroxynonenal (HNE), a major lipid peroxidation product [321,322,323,324,325]. Even though COX is viewed as the primary photoacceptor, molecular mechanism that elucidates the association of irradiation by 710–790 nm and 650–680 nm wavelengths with reduced and oxidized states of COX, respectively, remain elusive [326]. Furthermore, experimental work that combined nanoindentation and 670 nm laser irradiation to modulate viscosities of interfacial water supports the proposal that lower viscosity in mitochondria is the real driver behind photobiomodulation propelling enhanced ATP synthesis, and not increased COX expression and activities [327,328,329]. However, if reduced viscosity from light irradiation is responsible for increased ATP synthesis via increased power stroke velocity producing more efficient F1 motor rotations, then this proposal should be inclusive of COX involvement also.
3.2.3. Viscosity Modulates COX Activities in Mitochondria
In 1987, the main activity of COX—the oxidation of ferricytochrome C by COX—was demonstrated to be viscosity-dependent at both high and low ionic strengths [330]. While laser flash photolysis revealed a dramatic decrease in the rate of intramolecular electron transfer (IET) between the heme and molybdenum centers of chicken liver sulfite oxidase when solution viscosity was increased [331]. The evidence supporting the enhancement of ATP synthesis via light irradiation is solid [332,333], and it is also not unreasonable to propose that the reduction in mitochondrial matrix viscosity by light or ROS scavenging can increase ATP production, and increased ATP is associated with clearance of pathogenic aggregates from aberrant phase separation.
Thus, the ability to clear Aβ aggregation by the antioxidant epigallocatechin-3-gallate (EGCG) may be the result of upregulated COX activities and ATP production from reduced ROS and matrix viscosity [334,335]. In human neuroblastoma (SH-EP) cells, 670 nm irradiation dramatically elevated ATP levels by 20% which was subsequently diminished after irradiation-associated clearance of Aβ42 aggregation. Both 670 nm irradiation and EGCG were independently able to reduce Aβ42 aggregation at the expense of ATP consumption compared to controls. However, the combined, complementary treatment produced even better results in the clearance of amyloid aggregates compared to controls [334].
3.3. Melatonin Prevents and Disaggregates Aberrant Protein Aggregation in Dementia via Association with ATP
Melatonin, a mitochondria-targeted molecule [ 336 ] that is known for being a potent ROS scavenger [ 266 , 267 , 268 , 269 , 270 , 271 ], promotes ATP synthesis via the elevation of COX expression and activities. Melatonin administered to aged rats at 10 mg/kg per day in drinking water prevented the 30% age-related decline in COX activity while abolishing concomitant elevation of H 2 O 2 in brain mitochondria of aged rats compared to controls [ 337 ]. Melatonin administered orally at 10 mg/kg/day for 17 weeks to male Zücker diabetic fatty (ZDF) rats restored the 25% decline in renal mitochondrial COX activity and attenuated other mitochondrial dysfunctions including diminished ATP production compared to lean controls [ 338 ]. Melatonin administered in drinking water at the same amount to ZDF rats reversed the 76% decline in brown adipose tissue mitochondria COX activity by 35% while increasing COX activity by a staggering 31% in normal, lean controls [ 339 ].
3.3.1. Melatonin Elevates ATP Production via Modulation of COX and Viscosity
Even though cyanide (CN−)—a highly cytotoxic molecule that inhibits COX to suppress mitochondrial respiration and ATP production, and elevates ROS by modulating antioxidant defense—is proposed to be a novel mammalian gasotransmitter that can stimulate COX activity and enhance cellular bioenergetics at low endogenous nanomolar levels, at levels beyond 10 μM, CN− remains exceedingly toxic [340,341,342,343,344,345]. As a result, sophisticated dual-response sensors and probes are used to detect fluctuations in mitochondria viscosity in the presence of varying levels of cyanide in living cells [278,346].
Not unexpectedly, the in vitro study of rat brain mitochondria treated with 5 µM potassium cyanide revealed that 50% inhibition of COX activity was nearly entirely counteracted by treatment with 100 µM melatonin in a dose-dependent manner compared to control; while COX activity in rat liver mitochondria under same treatment conditions achieved 30% higher efficiency than control. However, at 100 µM cyanide exposure, even 5 mM of melatonin was unable to reverse the 100% inactivation of COX [347]. In vivo administration of melatonin at 10 mg/kg (IP) significantly elevated COX activity in rat brain and liver mitochondria in a time-dependent manner while reversing COX activity inhibition and preventing mitochondrial damage and oxidative stress induced by ruthenium red treatment at 60 µg/kg (IP) [348]. Ruthenium complexes can also increase viscosity and induce cell apoptosis via ROS-mediated mitochondrial pathways [349,350].
3.3.2. Fibril Disaggregation by Melatonin Is Dose-Dependent
Melatonin is intensively studied and extensively reviewed as a likely ideal therapeutic molecule for AD and other neurodegenerative disorders [ 351 , 352 , 353 , 354 , 355 ]. A novel understanding of melatonin regulation of biomolecular condensate phase separation in neurodegenerative disorders [ 230 ] provides additional relevant molecular mechanisms behind reported observation including the inhibition, destabilization, reduction, and delay of α-Syn and Aβ fibril aggregation. Melatonin not only increased survival rates in transgenic AD mice, but also reversed Aβ-induced synaptic disorder, memory deficit, neurodegeneration, as well as phosphorylation of tau in wild-type mice injected with Aβ peptides [ 356 , 357 , 358 , 359 , 360 , 361 , 362 , 363 , 364 , 365 , 366 ] ( Table 2 ). However, inconsistent results were observed when there were discrepancies in dosage and timing/duration of administration [ 363 ].
Table 2. In vitro and in vivo studies that reveal important relationships between melatonin dosage, timing, and administration that produced different results for symptoms associated with dementia.
| Melatonin Dosage/Duration | Study Design | Results | Ref. |
|---|---|---|---|
| 25 µM, 250 µM, 2.5 mM | In vitro α-Synuclein peptide aggregation | Blocked α-Syn fibril formation and destabilized preformed fibrils in a dose- and time-dependent manner; increased viability of primary mixed neurons treated with α-Syn to ~97% in a time-dependent manner. | [356] |
| 10 mg/kg (IP) ? 5/day for 2 days, then ? 2/day for 5 days | Arsenite-induced oxidative injury in substantial nigra of adult male rats | Attenuated arsenite-induced α-Syn aggregation, lipid peroxidation, and glutathione depletion. | [357] |
| 100 µM melatonin | A? peptides (1-40) and (1-42) ?-sheet/fibril formation | Progressive reduction in A?1-40 ?-sheet structures to 24% after 24 h incubation; immediate reduction in A?1-42 ?-sheet structures from 89% to 65%, decreasing to 59% after 4 h. | [358] |
| Melatonin dissolved in 2 mM ammonium acetate | A? peptide (1-40) ?-sheet/fibril formation | Inhibited ?-sheet formation by targeting hydrophobic A?-peptide segment (29-40) intermolecular activities. | [359] |
| 1 mM melatonin | A?1-40 peptide, profibrillogenic apoE4/apoE | Melatonin alone delayed fibril formation from 24 h up to 72 h. Combined with either apoE4 or apoE3, inhibition remained effective at termination of experiment. | [360] |
| 2 mg/mL in drinking water starting at age 4 months until euthanasia | Transgenic Tg2576 AD mice, terminated at 4 months 1 wk or 15.5 months | The brains of animals treated with melatonin terminated at 15.5 months exhibited dramatic decline in oligomeric A?40 together with a significant increase in soluble monomeric A?40, and a decreasing trend in A?42 compared to untreated mice at same age. Melatonin prolonged survival rates of 15.5-month mice to levels attained by non-transgenic mice. | [361] |
| 2 mg/mL in drinking water starting at age 4 months until euthanasia at 15.5 months | Transgenic Tg2576 AD mice | Increased survival in treated mice (3 deaths/41 survivals) compared to untreated (13 deaths/31 survivals). | [362] |
| 0.5 mg/mL in drinking water starting at age 4 months | Transgenic Tg2576 AD mice | Striking reductions in A? levels in brain tissues of treated mice at 8, 9.5, 11, and 15.5 months | [362] |
| 16 µg/mL in drinking water starting at age 14 months | Transgenic Tg2576 AD mice | Melatonin treatment failed to reduce brain A? levels or even oxidative damage. | [363] |
| 40-ppm (w/w) in pelleted minimal basal diet | Male B6C3F1 mice aged 6, 12, and 27 months | Significant reduction in A? in brain cortex tissues: 57% in A?40 and 73% in A?42; increased melatonin levels in cerebral cortex in all 3 treated age groups (12 > 6 > 27 mos) compared to untreated. | [364] |
| 10 mg/kg (IP) daily for 3 weeks | Male wild-type C57BL/6N mice (8 wks old) injected with A?1-42 peptide | Melatonin treatment reversed A?1-42-induced synaptic disorder, memory deficit, and prevented A?1-42-induced apoptosis, neurodegeneration, and tau phosphorylation. | [365] |
| 10 mg/kg in drinking water from day 7 after tauopathy induction to day 28 at termination | 4-month-old C57BL/6J mice injected with human tau mutation P301L (AAV-hTau) | MeIncreased ROS and tau hyperphosphorylation starting at day 7 precedes cognitive decline; melatonin-treated animals showed reduced memory impairment, tau hyperphosphorylation, ROS, and neuroinflammation.Increased ROS and tau hyperphosphorylation starting at day 7 precedes cognitive decline; melatonin-treated animals showed reduced memory impairment, tau hyperphosphorylation, ROS, and neuroinflammation.latonin treatment Increased ROS and tau hyperphosphorylation starting at day 7 precedes cognitive decline; melatonin-treated animals showed reduced memory impairment, tau hyperphosphorylation, ROS, and neuroinflammation. A?1-42-induced synaptic disorder, memory deficit, and prevented A?1-42-induced apoptosis, neurodegeneration, and tau phosphorylation. | [366] |
| 10 ?mol/L | Ex vivo brain slices from 3-month-old SD rats exposed to okadaic acid to induce tau hyperphosphorylation | Melatonin reduced tau hyperphosphorylation and ROS to control levels in OA-treated brain slices. | [366] |
| 100 ?M?5000 ?M | Aggregation/disaggregation of repeat domain Tau (K18wt) | Pre-formed tau fibril disaggregation was dose-dependent: 14% with 100 ?M, 54% with 5000 ?M. | [367] |
| 200?5000 ?M | Aggregation/Disaggregation full-length tau (hTau40wt) | Tau treated with 200 ?M melatonin showed no change in morphology compared to controls; 5000 ?M melatonin treatment did not prevent aggregation but disaggregated tau fibrils into broken filaments. | [368] |
Similarly, in vitro studies on tau fibril aggregation and disaggregation in the presence of melatonin at varying strengths found disaggregation effects to be dose-dependent where 100 μM led to 14% disaggregation while 5000 μM disaggregated ~54% of pre-formed repeat domain tau [367] (Table 2). However, 200 μM melatonin treatment in full-length tau aggregates failed to produce morphological changes, and 5000 μM treatment could not prevent aggregation but was able to disaggregate tau fibrils into small, broken filaments [368] (Table 2).
3.3.3. Melatonin Hydrogen Bonding May Modulate Salt Bridge Formation in Aggregates
The general consensus on the disaggregation mechanism employed by melatonin is the disruption of salt bridge formation or the reduction of hydrophobic interaction between proteins [358,359] (Table 2). Salt bridges formed between tau proteins can strengthen and stabilize the core of the paired helical filaments which enhances aggregation [367,369]. Both hydrogen bonds and salt bridges provide favorable free energy during protein–protein binding. Therefore, unfulfilled hydrogen bonds or isolated charges without forming salt bridges can destabilize binding due to the desolvation effect [370,371].
During Aβ oligomerization, the prerequisite expulsion of water molecules from protein hydration shells facilitates the formation of salt bridges [372]. In general, weaker hydrogen bonds are formed in interfacial regions due to the restrictive translational and rotational freedom constraints in interfacial regions. Consequently, more water molecules are required in interfacial regions for bridging hydrogen bond networks across protein interfaces [370]. When interacting with melatonin, water can act as both a H-bond donor to the amide carbonyl, methoxy oxygen, or indole π clouds and a H-bond acceptor from the amide NH and indole NH groups [252]. Therefore, the ability to form π hydrogen bonds [373] potentially allows melatonin to prevent the formation of salt bridges that impede intramolecular tau filament aggregation [374]. However, in vitro studies produced results that did not fully support in vivo and ex vivo work on melatonin and tau hyperphosphorylation [367,368] (Table 2).
3.3.4. Hyperphosphorylation Reduces Water Hydration during Fibril Aggregation
Hyperphosphorylation of tau is a reversible physiological process, but abnormal hyperphosphorylation in neurodegenerative disorders including AD is resistant to dephosphorylation and proteolysis [375,376,377,378]. It is believed that the cytotoxicity of Aβ is tau-dependent where tau and Aβ together drive healthy neurons into diseased states and that both Aβ and tau toxicity reinforce each other via a feedback loop [379,380,381,382]. The oligomerization of tau fibrils resulting in the formation of pathological tau aggregates is thermodynamically facilitated by hyperphosphorylation of tau proteins [383]. Hydrophobically driven phase separation which leads to the removal of water molecules from protein hydration shells is the predominant interaction that amplifies hydrophobic attractions that cause hyperphosphorylation of tau and fibrillization [135,384]. Recall tau proteins that phase separate due to salting-out effects mature into pathogenic, irreversible, canonical tau fibrils with restricted water accessibility and increased micro-viscosity [135] (see Section 2.3).
Furthermore, hyperphosphorylation can generate conformation changes critical for in vitro phase separation of full-length tau which precedes aggregation. Hyperphosphorylation shifts the equilibrium between soluble and phase-separated tau to favor the droplet state, enhancing maturation that initiates pathological aggregation [385]. Consequently, the ability to form hydrogen bonds to maintain protein solubility may determine the level of effectiveness of melatonin treatment in the prevention of tau hyperphosphorylation and subsequent phase separation events that ultimately result in the formation of pathological tau aggregates.
Therefore, in vitro work that showed dose-dependent disaggregation of pre-formed tau fibrils but the inability to prevent aggregation even at high concentrations of 5000 μM in contrast to in vivo and ex vivo work that reported a significant reduction in tau hyperphosphorylation even after the establishment of tauopathy (Table 2) may simply reflect the absence of ATP that can modulate hydrophobic interactions from hydrogen-bonding activities. In the context of phase separation in dementia, ATP may be the quintessential lynchpin that brings light, water, and melatonin together in a dynamic and effective synergy. After all, the regulation of aberrant protein aggregation in dementia by light and melatonin is associated with molecular mechanisms including reduced viscosity, hydrogen bonding, protein hydration, and elevation of ATP synthesis (Figure 1).
3.4. Light, Water, and Melatonin: The Adenosine Moiety Effect of ATP
The ability of ATP to solubilize hydrophobic substances in aqueous solutions at neutral and elevated pH was first reported by Mandl and Neuberg in 1952 [386]. Several decades later, ATP was observed to behave as a hydrotrope, solubilizing and dissolving protein aggregates in Xenopus oocyte nucleoli, preventing the aggregation of synthetic Aβ42 peptides, and even dissolving preformed tau fibrils [387,388]. However, employing all-atom molecular dynamics (MD) simulations, Kurisaki et al. observed contradictory results where ATP actually did not have any effect on the dissociation of monomers or the decomposition of the Aβ42 oligomer. Instead, the hydrophobic adenosine moiety of ATP was reported to dissociate Aβ42 monomers via contacts with Aβ42 backbone atoms, potentially dissolving the Aβ42 oligomer by shifting thermal equilibrium from an on-pathway species to an off-pathway species [389].
These observations were further clarified by Mehringer et al. demonstrating via MD simulations that ATP did not exhibit classic features of a hydrotrope or displayed chaotropic salting-in effects. In fact, ATP can be considered a kosmotropic anion with salting-out effects as a result of the triphosphate moiety of ATP capable of lowering the solubility of organic compounds in water. The ability of ATP to prevent and dissolve aggregates formed by phase separation observed in earlier works [387,388] is facilitated mainly by the interaction of the aromatic adenosine moiety in ATP with intrinsically disordered proteins, while the highly charged phosphate moiety served to heighten the solubility of the hydrophobic adenosine in ATP [390]. This molecular mechanism clearly explains why AD transgenic mice exhibit significantly reduced ATP production and mitochondrial dysfunction [391]. The adenosine moiety prevents hydrophobic collapse and aggregation by increasing solubility that prevents water removal [173]. As a consequence, the presence of ATP is highly effective in the suppression of Aβ16-22 peptide aggregation [392].
Adenosine is a primordial metabolite [393] that is an integral component of ATP and RNA [394,395]. Not unexpectedly, both ATP and RNA modulate phase separation biphasically where low concentrations enhance phase separation but high concentrations inhibit droplet formation [387,396,397,398,399]. MD simulations demonstrate succinctly that the dissolution of FUS by ATP-Mg2+ is promoted by solubilization via the adenine moiety and the phosphate moiety served only to enhance the requisite hydration effect [400].
Mechanistically, the adenosine moiety may prevent amyloid fibril formation by interfering with Aβ peptide π–π stacking [401,402]. Interestingly, the indole ring of tested indole derivatives effectively inhibited the formation of amyloid fibrils in hen egg-white lysozyme induced by low pH and high temperatures via hydrophobic interactions that accelerated disaggregation and destabilized the amyloid fibrillar state [403,404]. Therefore, it is perhaps not an evolutionary coincidence that melatonin not only exhibits structural homology to the adenosine moiety of ATP [401] (Figure 2), but also binds to adenosine via a hydrogen bond [405,406,407,408]. Consequently, ATP and melatonin may have been used for billions of years by living organisms to efficiently regulate phase separation in proteins with a high propensity for aggregation [230,401,409].
Arguably, the absence of ATP, despite the ability of melatonin to disrupt salt bridge formation, may be the reason why in vitro works on melatonin and tau fibril aggregation reported distinctly different results in the inhibition of fibril formation that could not confirm in vivo and ex vivo observations even at high concentrations of 5000 µM (Table 2).
Extracellular adenosine is derived from the degradation of ATP and adenosine monophosphate (AMP), whereas hydrolysis of AMP is the main source of intracellular adenosine [410,411]. It is estimated that extracellular adenosine can rise 1000-fold from the low nanomolar range of ~20–300 nM to the low micromolar range as high as 30 µM under conditions of high physical stress including extreme exercise and high altitude with low ambient oxygen [412]. Neurodegenerative diseases, inflammatory conditions, autoimmune diseases, cancer, diabetes, and cerebral ischaemia are pathological conditions associated with elevated extracellular adenosine [410,413,414,415,416,417].
Under optimal conditions, the high reserve/maximum capacity of melatonin synthesis in humans theoretically confers enhanced survival fitness as higher melatonin production allows rapid adaptation to unpredicted internal and external stressors [418]. Assuming that melatonin can be bound to adenosine at a 1:4 ratio [406,407,408], 6–20 nM plasma adenosine in venous blood collected from normal, healthy subjects [419] can theoretically bind to 1.5–5 nM of plasma melatonin. However, the lower range of 1.5 nM already reflects the highest 1.13 nM median level detected in nocturnal plasma melatonin concentration in children between the ages of 1–3 [420], and melatonin production begins to decline at the early age of 20–30 to approximately 0.12 nM after the age of 50 [421,422,423].
Furthermore, although there are contrary outcomes in some other reports, there may be conditions where endogenous production of melatonin is suppressed by constant exposure to 60 Hz magnetic field [424] and ambient light at night [425,426]. In addition to binding adenosine, melatonin can significantly elevate ATP production in mitochondria [347,348]. Therefore, the adenosine moiety effect of ATP in phase separation is directly affected by how much melatonin is available, and the dosage of melatonin becomes a critical moving target in the study of phase separation regulation in dementia.

4. Of Mice and Men: Perfecting the Human Equivalent Dose for Melatonin in the Regulation of Phase Separation in Dementia
The in vitro and in vivo effects of melatonin in dementia is not only dose-dependent, but may also be time-, and perhaps even age-dependent (Table 2). Three experiments that tested the same strain of transgenic Tg2576 AD mice with 0.016, 0.5, and 2.0 mg/mL of melatonin added to the drinking water starting at various ages, produced not only different, but also contradictory results (Table 2). Tg2576 mice are leaner compared to wild types as they age [430,431,432]. Assuming an average weight of 22.5 g for each animal drinking 3 mL of water per day [361,362], the approximate daily melatonin supplementation would have been 2.13, 66.66, and 266.66 mg/kg, respectively.
Tg2576 mice receiving ~2.13 mg/kg daily starting at age 14 months failed to show any benefit in the reduction in Aβ accumulation in the brain or oxidative stress levels [363]; whereas Tg2576 mice receiving ~66.66 mg/kg daily starting at age 4 months showed a significant reduction in Aβ levels in brain tissues, as well as lowered abnormal nitration of proteins [362]. Importantly, Tg2576 mice receiving ~266.66 mg/kg daily starting at age 4 months produced the most impressive results where the brains of mice terminated at 15.5 months not only exhibited a dramatic decline in oligomeric Aβ40, but also a significant increase in soluble monomeric Aβ40. A noticeable decreasing trend in Aβ42 was observed in treated compared to untreated mice at the same age [361]. When Tg2576 mice from two separate experiments were administered ~266.66 mg/kg melatonin in drinking water daily starting at age 4 months until termination at 15.5 months, survival was significantly increased in treated compared to untreated mice [361,362]. Melatonin treatment at ~266.66 mg/kg daily in drinking water was able to reduce mortality in Tg2576 mice to levels observed in wild-type mice [361] (Table 2). Consequently, the effective translation of melatonin doses between animals and humans becomes the primary consideration when designing the dosage for clinical trials.
4.1. Aiming at Moving Targets in Allometric Scaling of Melatonin Interspecies Conversion
Animals have different metabolic rates. In general, larger animals have lower metabolic rates; therefore, the metabolic rate requires scaling in the conversion of interspecies doses. Allometry broadly describes the study of consequences between body and organ sizes [433,434]. The concept of interspecies allometric scaling was first presented in 1637 by Galileo Galilei [435]. Since that time, various allometric approaches have been proposed and used to determine the most efficacious human equivalent dose (HED) [436,437,438,439,440,441,442,443,444,445,446,447,448,449,450]. However, the identification of a definitive unified principle that effectively scales and optimizes different energy metabolism systems across animal species remains elusive and highly controversial [441,451,452,453,454].
In 1880, Rubner first proposed the body surface law that scales metabolic rate with body mass raised to the power of ⅔ [455]. The seminal work by Kleiber in 1932 led to extensive empirical evidence that supports the metabolic rate in most animals and plants scales to the power of ¾ of body mass instead of ⅔ [454,456]. To date, there is no consensus as to whether ⅔ or ¾ power of body mass should be used as the basal metabolic rate scale in connection with the body mass to determine dose conversion. In general, the exponent of ⅔ may be more applicable for pharmaceuticals that are eliminated via the kidneys, whereas the exponent of ¾ is more suitable for molecules such as melatonin that are cleared by metabolism or via combined metabolism and renal elimination [441,445]. However, once a correction factor (Km)—a ratio that accounts for the interspecies difference between humans and animals obtained by dividing body weight by body surface area (BSA) [447,457]—is applied during the conversion of animal to human doses, where HED (mg/kg) = Animal dose (mg/kg) × (animal Km/human Km)
The results obtained are invariably higher than the ⅔ metabolic scale exponent of 0.67 (assuming human Km = 37.9).
For example, to calculate the HED for a 70 kg human, BSA 1.846, Km 37.9, the metabolic rate exponents for a mouse weighing 0.02 kg, BSA 0.007 (M2), Km 2.857, and Km ratio 13.265; and a rat weighing 0.15 kg, BSA 0.025 (M2), Km 6, and Km ratio 6.317, after adjustments would be 0.683 and 0.700, respectively. For a mini pig weighing 40 kg, BSA 1.14 (M2), Km 35.088, and Km ratio 1.08, the exponent for metabolic scaling for a 70 kg human becomes 0.863.
Thus, the accuracy of HED calculations during interspecies conversion is strictly dependent upon both body weight and BSA. The human BSA can be estimated via the formula BSA = ⅙ (Weight × Height)0.5 [458], whereas specific animal BSA is often more difficult to ascertain. Popular manuals containing instructions for interspecies HED conversions rely upon predetermined animal BSAs based upon a mathematical formula (BSA = kW2/3) postulated by Meeh in 1879, where BSA is derived from a constant k and volume estimated from body mass that is scaled to the ⅔-power. For most small animals including mice, the mean constant k is accepted to be 9.83 [459,460]. However, empirical determination for Meeh constants in mice with different body compositions and shapes revealed a range from 9.822 (normal) to 8.288 (obese) [459]. Therefore, the difference in Meeh constants between measured and calculated values would be important considerations when using mice with altered body composition. Furthermore, modifications for cell porousness per fractal theory indicate that the scaling exponent can vary from 0.694 to 0.83 [450].
As such, the estimated HEDs in this review will be calculated employing metabolic rate scaling using body weight raised to the ¾-power where
HED (mg/kg) = animal dose (mg/kg) × (WEIGHT[kg]animal/WEIGHT[kg]human)(1−0.75)
Using this formula, the estimated HED for a rat weighing 0.2 kg with a 10 mg/kg melatonin dose will be 2.12 mg/kg and 2.4 mg/kg for a 100 kg and 60 kg human, respectively. Nevertheless, the often-large differential in interspecies bioavailability and pharmacokinetics that can be modulated by route of administration, dosage, solubility, and formulation must also be taken into account for the accurate determination of an efficacious HED during conversion/scaling processes.
4.2.1. Administration Routes Modulate Melatonin Bioavailability
The bioavailability of melatonin is affected by different routes of administration, where the mean bioavailability for 25 mg of melatonin delivered via intravesical, transdermal, rectal, and vaginal administration in healthy female volunteers were 3.6%, 10.0%, 36.0%, and 97.8%, respectively, compared to IV administration [463]. However, the determination of the bioavailability of oral melatonin may be complicated by melatonin metabolism. In humans, melatonin is mainly cleared by first-pass hepatic metabolism. When the clearance of an IV melatonin dose was combined with plasma concentrations of oral doses from previous data, the calculated oral bioavailability of melatonin was estimated to be 3–6% after a 2.5 mg dose, 3–76% after an 80 mg dose, but only 9% after a 100 mg dose [464].
In 2000, DeMuro and coworkers determined the absolute oral bioavailability of 2 and 4 mg melatonin doses (tablets) to be 14.3% ± 7% and 15.9% ± 6%, respectively, compared to IV melatonin (2 mg) [465]. Fifteen years later, a systematic review of 22 studies identified from 392 records that tested oral or IV melatonin dosages between 0.3 and 100 mg found the bioavailability of melatonin to be approximately 15% with significant variability between individuals, where critically ill patients often displayed accelerated absorption but compromised elimination [466].
4.2.2. Melatonin Bioavailability Is Inversely Correlated to 6-Sulfatoxymelatonin
As such, the interpretation of bioavailability may not be straightforward considering the fact that absolute bioavailability of oral melatonin has also been reported at ~3% (10 mg gelatin capsule) albeit with considerable variability among the 12 tested healthy male subjects (20–40 yr old) [467]. Low absolute bioavailability in oral melatonin is often the prominent effect of first-pass hepatic metabolism which produces the major melatonin metabolite 6-sulfatoxymelatonin (6-OHMS). Consequently, low endogenous production of melatonin in the elderly is associated with a significant reduction of 6-OHMS in older test subjects compared to younger ones (82–21 years old) [421]. However, a small study sample found a significant inverse correlation between oral bioavailability and 6-OHMS, where lower oral bioavailability (10%, 12%) was correlated with high plasma 6-OHMS (31%, 14%). Conversely, high bioavailability (56%, 54%) was associated with lower 6-OHMS in plasma (4%, 3%) of healthy male subjects (21 to 32 years old) tested [468].
This inverse relationship was also observed in children admitted to an intensive care unit where septic patients who did not survive exhibited nocturnal melatonin levels that were significantly higher than survivors, but total 6-OHMS excretion was dramatically lower in nonsurvivors compared to survivors. Additionally, septic shock patients had higher nocturnal melatonin levels than non-septic patients [469]. Low plasma 6-OHMS is correlated with autism [470,471], and low 6-OHMS excretion level is associated with adults who were lean at birth but obese in adult life, and high excretion rates were associated with opposite observations [472]. Similarly, patients with unstable angina exhibited significantly lower 6-OHMS than healthy controls and no negative correlation with age was observed in coronary patients as opposed to healthy subjects [473]. Therefore, the interpretation of melatonin bioavailability becomes more meaningful when 6-OHMS levels are taken into consideration.
4.2.3. Animals Show Large Variations in Melatonin Bioavailability
In animals, melatonin bioavailability via different administration routes varies greatly with strain, species, and first pass metabolism after administration. Yeleswaram and coworkers determined the absolute bioavailability of melatonin for a 10 mg/kg oral dose compared to IV in male Sprague Dawley (SD) rats to be 53.5%, but more than 100% in dogs and monkeys. However, the oral bioavailability in dogs is dose-dependent, where 1 mg/kg resulted in only 16.9% bioavailability. IP injection of melatonin at 10 mg/kg in SD rats increased bioavailability to 74.0% compared to oral at 53.5%.
In rats, IV administration at half the dose (5 mg/kg) achieved 80% bioavailability via IP at 10 mg/kg [474]. Rats, regardless of strain and administration, metabolize melatonin completely. SD rats excreted 60–70% of radiolabeled melatonin via IV injection as the major metabolite 6-OHMS [475]; and male Wistar rats administered 12.5–250 µg melatonin via IP injection also showed concentration-dependent increases in plasma of melatonin and 6-hydroxymelatonin, which always maintained a constant ratio of 1% of plasma melatonin irrespective of dosage. However, the sulfate conjugate 6-OHMS reached at maximum, ~64-fold elevation of maximum plasma 6-hydroxymelatonin levels [476].
Similar to rats, female C57BL/6 mice (age 8–10 weeks) administered varying doses of melatonin at 31.25, 62.5, 125, 250, and 500 mg/kg showed no difference in the ability to clear and eliminate melatonin; and the concentration of melatonin in the liver and gastrointestinal tracts was higher than other vital organs by 5- to 10-fold, indicating that hepatic first-pass metabolism is also prominent in mice. However, the effect of melatonin in mice is also dose-dependent even at supra-pharmacological concentrations. After exposure to lethal radiation, mice administered 500 mg/kg had the highest survival rate (55%) compared to 250 and 125 mg/kg (40%) [477].
4.2.4. Solubility and Formulation Modulate Melatonin Bioavailability
The oral bioavailability of melatonin, at any dose, can be modulated by altering solubility. The oral bioavailability of melatonin in critically ill patients with sepsis was greatly enhanced by the use of solvents, where melatonin dissolved in glycerol achieved a 5-fold increase in relative bioavailability over melatonin in capsules at the same doses (20 or 50 mg) [478]. Similarly, when compared to IV solution (62.5 mg/kg dissolved in water), absolute oral bioavailability in mice of an aqueous melatonin suspension at 250 mg/kg administered via gavage tube was 29%, whereas 250 mg/kg melatonin dissolved in a popular co-solvent polyethylene glycol 400 (PEG400) and administered in the same manner achieved absolute bioavailability of 98.5% [477]. However, PEGs are very hydrophilic molecular crowders that can amplify entropy gain from water-release, causing dehydration that drives phase separation [136]. Hence, the use of PEG as a solvent in applications for the regulation of phase separation must be carefully weighed.
Variations in formulation also affect melatonin bioavailability. In rabbits, intranasal delivery of melatonin encapsulated in starch microspheres achieved absolute bioavailability of 84.07%, whereas intranasal administration via solution produced much lower pharmacokinetics [479]. While intranasal melatonin administration in male Wistar rats via niosomes—bilayer vesicles of nonionic surfactant-based liposomes—achieved absolute bioavailability of 98.7% compared to IV melatonin solution [480]. Therefore, the successful conversion of an animal melatonin dose into an efficacious human equivalent requires equal considerations of metabolic rate scaling, bioavailability as determined by intrinsic differences between species, administration route, as well as solubility and formulation.
4.3. Timing Is Everything in the Dosing of Melatonin for the Regulation of Phase Separation in Dementia
The daily supra-pharmacological dose of 266.66 mg/kg administered in drinking water to transgenic Tg2576 mice from 4 months to 15.5 months not only prevented aggregation of amyloid fibrils but also prolonged survival compared to untreated mice [361,362]. This dose can be converted into a HED using metabolic scaling to the ¾-power, with the assumption of mice and human body weight to be 0.0225 kg and 70 kg, respectively; and oral bioavailability of mice and humans to be 63.75% and 15%, respectively. 63.75% oral bioavailability is a conservative estimation of a 50% enhancement of solubility in water achieved by first dissolving melatonin in hydroxy methyl cyclodextrin before dilution in drinking water to the final concentration of 2 mg/mL [361]. Cyclodextrins (CDs) are small carbohydrates that enhance the solubility of molecules and drugs, resulting in higher bioavailability [481,482]. The HED obtained before the bioavailability adjustment is 2499 mg. Without solubility enhancement, the adjusted bioavailability HED dose is 4831 mg. After correcting for a 50% enhancement in bioavailability (calculated based on oral bioavailability data obtained by Choudhary et al. [477]), the final HED is a staggering dose of 10,621 mg.
Even though the toxicity of melatonin as defined by LD50 has not been determined in human or rodents, where early studies failed to produce death in mice at 800 mg/kg [483], and acute oral toxicity that result in LD50 in rats is reported at concentrations higher than 3200 mg/kg (in one single dose [484]) according to the latest Merck safety data sheet on melatonin (Regulation (EC) No. 1907/2006, revised 17 November 2021), without a convincing rationale for a high HED in the context of phase separation in dementia, this extreme supra-pharmacological HED may seem unjustified.
4.3.1. The Rationale for Frequent Division of Melatonin Doses
The Tg2576 mice drank ~3 mL of water containing a total of 6 mg melatonin in a 24 h period. Accordingly, total HED should also be administered in divided doses of 885 mg × 12. This hypothetical HED now resembles the HED used by Martin et al. to elevate ATP production via complex I and COX (complex IV) activities in the brain and liver mitochondria of rats [348].
Male Wistar rats with a body weight between 200–230 g were administered 10 mg/kg melatonin via IP injection. Respiratory complex activity enhancements were tissue- and time-dependent. Complex I activities in the liver achieved peak levels and returned close to baseline at ~30 and ~180 min, respectively; whereas in the brain, peak activity levels were attained at ~60 min and returned to baseline at ~180 min. COX activities in both the liver and brain became significantly elevated at ~30 min, but reached a peak in the liver at ~100 min before declining close to baseline at 180 min, whereas brain activities quickly dropped to baseline soon after 120 min [348]. In other words, in the brain, complex I and COX reached peak activity levels at 60 and 30 min, respectively, before returning to baseline at ~120 min. Whereas in the liver, complex I and COX activities were both elevated at ~30. Complex I steadily declined to close to baseline at ~180 min, but COX activities remained elevated and reached a peak at ~100 min before declining close to baseline at ~180 min.
The difference in peak and duration of respiratory enzyme activities may reflect the effect of prominent first-pass hepatic metabolism where more melatonin is retained in the liver and gastrointestinal tracts than other vital organs such as the brain [475,477]. Regardless, bioavailability via IP in rats is ~74%, or 4.933-fold higher than oral bioavailability in humans. Therefore, assuming an average body weight of 0.215 kg and 70 kg for rats and humans, respectively, the total daily HED that can effectively maintain peak complex I and COX activity at a sustained level throughout a 24 h period in order to provide adequate ATP and adenosine that can prevent and solubilize aberrant phase separation and aggregation is 9755 mg, adjusted for differences in metabolic rate and bioavailability, assuming average intake of 812.90 mg × 12 in a 24 h period. However, the amount quickly doubles to 19,509.60 mg if maximum complex I and COX activities were to be sustained in the brain over a 24 h period based on observations reported by Martin et al. [348].
At this point, the estimated daily total HED of 10,621 mg obtained from Tg2576 mice taking 266.6 mg/kg in drinking water becomes quite reasonable and theoretically justifiable. In addition, in vitro work found a strong correlation between ATP and melatonin concentration for disaggregation of fibrils [367,388], where 1 mM of ATP and melatonin both were able to dissolve 20% of aggregates, respectively; 4 mM and 5 mM of ATP and melatonin dissolved 50% and ~60% of aggregates, respectively (Figure 3). Therefore, the rationale supporting supra-pharmacological oral melatonin doses to maintain elevated ATP synthesis that prevents aberrant phase separation and aggregation warrants further investigation.

4.3.2. The Calculation of HED Estimates Adjusted for Differences in Metabolic Rates, Bioavailability, and Formulation
A close examination and comparison of various HED estimates obtained from the different in vivo experiments discussed in Table 2 may provide clarification on melatonin doses required for the effective regulation of phase separation in dementia. Importantly, there is a difference in doses required to obtain similar results in healthy versus diseased, transgenic animal models.
Table 3 illustrates how (A) oral melatonin HED for a human weighing 70 kg is converted from animal doses by using metabolic rate scaled to ¾ power with body weight (Mb3/4); (B) where HED (A) is further adjusted by interspecies bioavailability difference that takes into account both species differentials and administration routes; and (C) adjusts (A) to reflect enhancements via solubility/formulation as per study design. In the absence of data, where applicable, the average body weight of transgenic Tg2576 and wild-type mice is assumed to be 0.0225 kg and 0.025 kg, respectively. Daily food intake for mice [364] is assumed to be ~4.5 g [432]. The oral bioavailability of melatonin in humans and mice is assumed to be 15% [465,466] and 29% [477], respectively, in the conversion for values in column (B). Bioavailability enhancement via increased solubility is estimated at a conservative 50% increase based on data reported by Choudhary and coworkers [477]. Therefore, values in column (C) are obtained by multiplying (A) by 4.25.
| tudy Design/Total Daily Dose/Duration/Ref. | Results | (A) HED Daily Total (mg/kg) Scaled to Mb3/4 | (B) Dose (A) Adjusted by Bioavailability | (C) Dose (A) Adjusted by Enhanced Bioavailability |
|---|---|---|---|---|
| 2 mg/mL in drinking water, Tg2576 AD mice/266.66 mg/kg/11.5 mos starting at 4 mos old/[361,362] | Striking reductions in A? aggregates at all ages during treatment; dramatic extension of survival of AD mice to levels similar to wild types. | 2499 mg (35.7 mg/kg) | 4831 mg (69 mg/kg) | 10,621 mg (151.73 mg/kg) |
| 0.5 mg/mL in drinking water, Tg2576 AD mice/66.66 mg/kg/11.5 mos starting at 4 mos old/[362] | Striking reductions in A? levels in brain tissues of treated mice at 8, 9.5, 11, and 15.5 months. | 625 mg (8.928 mg/kg) | 1208 mg (17.26 mg/kg) | 2656 mg (37.94 mg/kg) |
| 0.016 mg/mL in drinking water, Tg2576 AD mice/2.13 mg/kg/10 wks starting at age 14 mos old/[363] | Failed to reduce brain A? levels, unable to reverse oxidative damage. | 19.96 mg (0.285 mg/kg) | 38.58 mg (0.55 mg/kg) | 84.83 mg (1.21 mg/kg) |
| 10 mg/kg in drinking water, healthy, normal C57BL/6J mice/14 days after tauopathy initiation/[366] | Reduced memory impairment, tau hyperphosphorylation, ROS, and neuroinflammation. | 96.23 mg (1.375 mg/kg) | 186.0 mg (2.66 mg/kg) | 408.98 mg (5.84 mg/kg) |
| 40 ppm in food pellets, healthy, normal B6C3F1 mice/7.2 mg/kg/11 weeks different age groups/[364] | Significant reduction in A? peptides in brain cortex tissues: 57% in A?40 and 73% in A?42; increased melatonin levels in cerebral cortex in all 3 treated age groups (12 > 6 > 27 mos) compared to untreated. | 69.29 mg (0.99 mg/kg) | 133.94 mg (1.91 mg/kg) | Not applicable |
| 10 mg/kg IP injection, C57BL/6J mice treated with A?1-42 peptide/daily IP injections for 3 wks/[365] | Reversed A?1-42-induced synaptic disorder, memory deficit; prevented A?1-42-induced apoptosis, neurodegeneration, and tau phosphorylation. | 98.55 mg (1.41 mg/kg) | 486.15 mg (6.95 mg/kg) | Not applicable |
The selection of a “perfect” HED dose for melatonin under different contexts is ultimately at the sole discretion of the investigator(s) who will determine the “parameters to be scaled, independent variables, and the mathematical relationship used in the scaling process” [438,439]. Therefore, the values presented in Table 3 are intended purely as an informative guide to various potentially effective HEDs for melatonin that can be applied in the regulation of phase separation in dementia under distinct conditions.
5. Conclusions
Modernization of infrastructure leads to inevitable environmental changes that restrict easy access to the ancient, dynamic synergy between light, water, and melatonin. Individuals who live in densely populated urban areas are affected by the lack of adequate greenness that limits exposure to red and infrared frequencies from sunlight, generously reflected by plants [205]. Furthermore, continuous exposure to low-level microwaves and varying levels of EMF can restructure hydrogen bonding to either decrease or increase intracellular viscosity [195,196,197,198,199]. Even exposure to magnetic fields at 0.5 T causes water molecules to form new hydrogen bonds resulting in larger-sized water clusters that increase viscosity but reduce the proportion of free water molecules [485]. At the same time, the endogenous production of melatonin may be impacted under some circumstances by constant exposure to 60 Hz magnetic field [424] and ambient light at night [425,426]. In older adults with varying risks for dementia, increased light exposure in the evening results in earlier dim light melatonin onset (DLMO) time. This shift in the circadian phase may disturb rhythmicity that is often associated with dementia [486,487,488].
Our brave, new world offers unlimited potential in technological advances in every frontier imaginable but exacts an exorbitant premium on our health by creating intracellular conditions that favor aberrant phase separation resulting in pathological protein aggregations that are associated with a wide range of health challenges, including dementia. The reinstatement of this powerful but lost synergy is a provocative proposal that entails the conditional rescaling of an ancient theme to harmonize with the cacophony of modern influences, restoring, once again, balance in optimum health.
Author Contributions
- D.L.: Conceptualization and manuscript preparation. R.J.R.: Critical review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
- This research received no external funding.
Institutional Review Board Statement
- Not applicable.
Informed Consent Statement
- Not applicable.
Data Availability Statement
- Not applicable.
Acknowledgments
- Special thanks to Daniel Matrone for technical assistance. Figure 1 was created with BioRender.com.
Conflicts of Interest
- The authors declare no conflict of interest.